KOLCHANOV N.A., PONOMARENKO M.P., KONDRAKHIN Y.V., FROLOV A.S., KOLPAKOV F.A., KEL A.E., KEL-MARGOULIS O.V., ANANKO E.A., IGNATIEVA E.V., PODKOLODNAYA O.A., STEPANENKO I.L., MERKULOVA T.I., BABENKO V.N., VOROBIEV D.G., LAVRYUSHEV S.V., GRIGOROVICH D.A., PONOMARENKO J.V., KOCHETOV A.V., KOLESOV G.B., PODKOLODNY N.L.1, WINGENDER E.*, HAINEMEIER T.*, MILANESI L.2, SOLOVYEV V.V.3, OVERTON G.C.4
Institute of Cytology and Genetics, (Siberian Branch of the Russian Academy of Sciences), 10 Lavrentieva ave., Novosibirsk, 630090 Russia; 1Institute of Computational Mathematics and Mathematical Geophysics, SB RAS, Novosibirsk, 630090 Russia; *Gesellschaft fur Biotechnologische Forschung mbH, Mascheroder Weg 1, D-38124 Braunschweig, Germany; 2ITBA CNR, Milan, Italy; 3The Sanger Centre Hinxton, Cambridge, CB10 1SA, UK; 4Center for Bioinformatics, University of Pennsylvania, 2323 Blockley Hall, Philadelphia, PA 19104-6021, USA
Keywords: eukaryotic genomes, regulatory sequences, database integration, knowledge discovery
Currently, the number of databases on gene expression and variety of software for the analysis of these data are growing fast. That is the reason why the following problems of the modern bioinformatics of gene expression regulation are challenging: (a) creation of the unified Internet-accessible media to provide maximal integration of information-software resources on gene expression and effective navigation of users in the integrated media; (b) development of a technique for automated discovery of knowledge on structural and functional organization of regulatory genomic sequences and on their recognition; (c) activation of databases on regulatory genomic sequences and their conversion from passive information carriers to the active information and software modules to provide the users with various active knowledge, for example, such as the programs for analysis and recognition of regulatory genomic sequences. The system GeneExpress [1] is developing to pursue these goals. It includes five basic units: (1) Transcription Regulation unit contains database on transcription regulatory regions of eukaryotic genes [2]; (2) Site Recognition unit contains programs for analysis and recognition of functional sites [3, 4]; (3) ACTIVITY is the module designed for site activity prediction from their nucleotide sequences [5-8]; (4) mRNA Translation unit is designed for analysis of translational properties of mRNAs [9]; (5) GeneNet is the database for accumulation of the data on eukaryotic gene networks and signal transduction pathways [10,11]. All the units are integrated into the GeneExpress system on the basis of the Network Browser of Sequence Retrieval System (SRS).
The core element of GeneExpress is TRRD: Transcription Regulatory Regions Database [2]. TRRD describes the modules of transcription regulatory regions and the hierarchy of their organization: cis-elements, composite elements, promoters, enhancers, silencers, and the extended transcription regulatory regions. It provides description of different features of transcription regulation for example its dependence on the cell cycle stage, developmental stage, tissue-specificity, or effects of external factors, etc. It is linked to the relevant databases on transcription regulation (TRANSFAC, EPD, EPODB, COMPEL, GERD) as well as to various software for analysis of regulatory genomic sequences included into the GeneExpress system. At present, GeneExpress contains the description of more than 400 genes including over 2000 transcription factor binding sites. The most detailed description is given for the cell cycle gene family, erythroid – specific genes, genes involved in lipid metabolism, interferon-inducible genes, glucocorticoid-regulated genes, muscle-specific genes, and some others [12-16]. Information from TRRD could be visualized by the Java applet TRRD Viewer [17].
There is a possibility for user to move from TRRD to a particular knowledge base of the GeneExpress system via appropriate links (Figure 1). Among the currently available knowledge bases of GeneExpress are the following: knowledge bases on significant conformational, physico-chemical properties of DNA sites [1, 18], knowledge base on functional sites recognition [1,4], and knowledge base on functional site activities [1,6.8]. All this knowledge is discovered automatically by the special tools of the GeneExpress system.
System of knowledge discovery on recognition of functional sites permits to generate programs for functional sites recognition on the basis of consensuses and weight matrices [1,4]. The basic units of this system are: (1) Samples database, containing functional site sequences [19]; (2) database of functional site alignments [19]; and (3-4) modules for automated generation of recognition programs by consensuses and weight matrices [1,4]. Compilation of the sets of functional sites included into the Samples database is performed by an object-oriented system MGL (Molecular Genetic Language) [17], which provides automated extraction of regulatory genomic sequences on the basis of semantic analysis of information from EMBL, TRRD, TRANSFAC, and EPD databases. Automatically generated C-codes of recognition programs are stored in the knowledge base with indication of the overall recognition error rates [4]. Figure 2 demonstrates a fragment of the C-code of program generated automatically to recognize the C/EBP-transcription site by using its consensus of the trinucleotide alphabet {MMM,MMK,MKM,MKK,KMM,KMK,KKM,KKK} at each position of this site DNA sequences.
On the basis of the same set of sequences, dozens of different programs to recognize a particular site are generated automatically. To use all this totality of the methods stored in the knowledge base, we have developed the technique named the mean recognition [4]. It is based on the fundamental fact that summarizing the results of site recognition on the basis of a large number of programs leads to systematic increase in the accuracy rate [4]. Mean recognition significantly decreases both the type I and II errors, a1 and a2, with respect to the conventionally used methods of consensus and weight matrix.
To obtain the knowledge about the conformational and physico-chemical properties of the site, a molecular biologist can use the B-DNA-VIDEO system [18, 20]. In addition to the above-described database Samples [19], B-DNA-VIDEO includes: (1) the database on context-dependent DNA conformational and physico-chemical properties; (2) the unit for automatic knowledge discovery on significant conformational and physico-chemical properties of the sites; and (3) the Internet-accessible knowledge base containing the results of analysis. It is well known that local conformational and physico-chemical properties of B-DNA double helix are context-dependent [21]. Currently, the GeneExpress system contains description of more than 30 context-dependent B-DNA conformational, physico-chemical parameters]. The automatic knowledge discovery system [18,20] searches for the site regions [a,b], within which the mean value of the parameter in question Xk,a,b(S) differs significantly from the corresponding value calculated for random sequences Xk,a,b(R). The discriminative ability of the characteristic Xk,a,b is estimated in terms of mathematical variable called utility, Uk,a,b, using approaches of the fuzzy logic applied within the framework of the Utility Theory for Decision Making [22]. Utility has the following properties: (1) it varies in the range of -1 to +1; (2) only the characteristics Xk,a,b with positive utility can be used for discrimination of sites from nonsites; (3) the larger is the utility value Uk,a,b, the more efficient is the characteristic X for discriminating sites from nonsites. Only a limited set of linearly independent characteristics with the maximal utility is selected for each site. For example, En (Engrailed) site is characterized by the decreased value of conformational property ROLL angle within the region [-10;2] (Figure 3a ). In this case, the distribution of the mean value of ROLL for sites is right-shifted significantly in comparison with that for random sequences. As for HNF1 site, the most important is the decreased DNA melting temperature in the region [-21,4] (Figure 3b). Analysis of transcription factor binding sites has demonstrated that the majority of them are characterized by specific sets of conformational and physico-chemical properties (for details, see [18,20]. Information on the significant conformational and physico-chemical properties of the sites is stored in the knowledge base of the B-DNA-VIDEO system [18,20]. It contains also the references to the clickable programs for constructing the profiles of significant conformational and physico-chemical properties of sites and searching for the regions that are maximally similar in this property to the actual functional sites. The C-codes of relevant programs are also available.
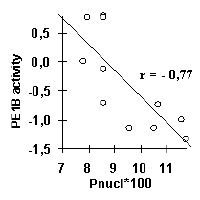
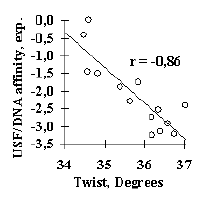
The molecular biologist interested in investigating specific site activity can further proceed to the ACTIVITY system [1,5-7]. It includes: (1) the database on functional site activities; (2) automatic knowledge discovery system for revealing contextual, conformational, and physico-chemical properties significant for the site activity and (3) Internet-accessible knowledge base compiling the results of analysis. Currently, more than 400 sets of functional sites with experimentally determined levels of their specific activity are described in the ACTIVITY system [1,5-7]. The knowledge discovery system applies multiple regression model to contextual, conformational, and physico-chemical properties for prediction of the site activity [1,5-7]. Some results obtained by the knowledge discovery system are shown in Figure 4. For example, the USF/DNA affinity correlates very well with the helical twist of B-DNA; the major groove width determines the Cro repressor/DNA affinity; transcriptional activity of the mouse alphaA-crystalline gene promoter is determined mainly by the rate of contacts with nucleosome core proteins. Conformational and physico-chemical properties significant for activity prediction have been revealed for most of the sites considered. They are stored in the knowledge base of the ACTIVITY system together with the Internet-accessible programs for prediction of site activities from their nucleotide sequences [1,5-7].
GeneExpress system is also supplemented with a special tool SeqAnn for activating the TRRD database while revealing putative promoters [23]. The core idea of this approach is to use the information from the TRRD database as a ready-to-use scenarios for promoter recognition. In a simple case, it allows a user to search a target sequence for putative promoters which are similar to actual promoters described in TRRD basing on the content and approximate location of transcription factor binding sites. In the pilot release of SeqAnn [23], the search for sites is performed by the homology approach. At the next stage, the entire set of the site recognition methods accumulated in GeneExpress knowledge bases will be employed. And finally, as it was noted above, the simultaneous use of a large number of recognition programs for individual sites will rise the accuracy of recognition. The information on the activity of the site to be recognized will be also used for the same purpose.
The GeneNet database [10,11], a constituent of the GeneExpress system, describes the highest level of integration responsible for the hierarchical organization of gene expression regulation. GeneNet is designed within the object-oriented approach to description of experimental data on gene networks and signal transduction pathways. One more goal of GeneNet is to provide additional possibilities for navigation within GeneExpress. It supports the cross-references with EMBL, SWISS-PROT, TRRD, TRANSFAC, and EPD databases. The current release of the GeneNet database [10,11] contains the descriptions of gene networks of interferon-inducible and erythroid-specific genes. The three following hierarchical levels are considered: (1) organism level, (2) the single cell level, and (3) the single gene level, using the data on transcription regulation from the TRRD database. A special language for the formalized description of gene network events is being developed. A fundamental advantage of this approach is automated visualization of the database entries [10,11]. Further development of the GeneExpress system will include: (1) the development of all database and software modules currently available; (2) creation of new techniques for automatic knowledge discovery systems; (3) development of new approaches for activation of databases; and (4) integration of new external database and software resources.
This work is supported by grants from the Russian Foundation for Basic Research (No.97-04-49740, 97-07-90309, 96-04-50006, 98-04-49479, 98-07-90126); Russian Ministry of Science and Technologies; Russian Human Genome Project; Russian Ministry of High Education; Siberian Department of RAS (Programms for support of reseach of young scientists and Programm of Integration projects); National Institutes of Health, U.S.A. (No.5-R01-RR-04026-08)
References
- Kolchanov N. A. et al., (1998) Genexpress: a computer system for description, analysis, and recognition of regulatory sequences of the eukaryotic genome. Proceedings of the Sixth International Conference on Intelligent Systems for Molecular Biology, AAAI Press, Menlo Park, California, USA, 95-104.
- Kel’ A.E. et al., et al., (1997) TRRD: database on transcription regulatory regions of eukaryotic genes. Mol. Biol. (Msk) 31, 521-530.
- Kondrakhin Yu.V. et al., (1998) Recognition groups: a new method for description and prediction and of transcription factor binding sites. CABIOS (in press)
- Ponomarenko M.P. et al., (1998) Recognition accuracy of DNA functional sites can be increased by averaging partial recognitions. Proc. of the 1st BGRS Conference, Novosibirsk, ICG, SD RAS, this issue
- Kolchanov N.A. et al., (1998a) Functional sites in pro- and eukaryotic genomes: computer models for predicting activity. Mol. Biol. (Mosk), 32, 2, 255-267.
- Ponomarenko J.V. et al., (1998) ACTIVITY: A database for activities of functional DNA/RNA sites. Proc. of the 1st BGRS Conference, Novosibirsk, ICG, SD RAS, this issue.
- Ponomarenko M.P. et al., (1997).Generating programs for predicting the activity of functional sites. J. Comput. Biol., 4, 83-90.
- Ponomarenko M.P. et al., (1997a) A distributed and intelligent database for the activities of the functional sites in DNA and RNA. Abstracts of the 1998 Pacific Symposium on Biocomputing. Ed.: Altman et al., World Scientific, P. 103-104
- A.V. Kochetov et al., (1988) Structural and compositional features of 5’-untranslated regions of higher plant mRNAs. Proc. of the 1st BGRS Conference, Novosibirsk, ICG, SD RAS, this issue
- Kolpakov F.A. et al., (1998), GeneNet: a database for gene networks and its automated visualization through the Internet. Bioinformatics, in press.
- Ananko E.A. et al., (1988) GENE NETWORKS: a database nd its automated visualization through the internet in the GeneNet system. Proc. of the 1st BGRS Conference, Novosibirsk, ICG, SD RAS, this issue.
- Ignatieva E.V. et al., (1997) Transcription regulation of lipid metabolism genes as described in the TRRD database. Mol Biol (Mosk) 1997. V 31. p 575-591
- Anan`ko E.A. et al., (1997) Mechanisms of transcription of the interferon-induced genes: a description in the IIG-TRRD information system. Mol Biol (Mosk) 1997, v. 31, 592-605.
- Merkulova T.I. et al., (1997) Mechanisms of glucocorticoid regulation and regulatory regions of genes, controlled by glucocorticoids: description in the TRDD database. Mol Biol (Mosk) 1997, v.31, p. 714-725.
- Kel OV, Kel AE (1997) Intergenic interrelations in regulating the cell cycle: key role of E2F family transcription factors. Mol Biol (Mosk) 1997, v.31, p. 656-670.
- Podkolodnaia OA, Stepanenko I.L. (1997) Mechanisms of transcriptional regulation of erythroid specific genes. Mol Biol (Mosk) 1997, v.31, p. 671-683.
- Kolpakov, F.A. and Babenko, V.N., 1997, Computer system MGL: tool for sample generation, visualization and analysis of regulatory genomic sequences. Mol. Biol. (Mosk), v.31, p. 540-547.
- Ponomarenko M.P. et al. (1998) Revealing the conformational and physico-chemical DNA features for predicting the activity of the functional sites, Proc. of the 1st BGRS Conference, Novosibirsk, ICG, SD RAS, this issue.
- Vorobiev D.V., Ponomarenko JV (1998), SAMPLES and ALIGNED: databases for functional site sequences. Proc. of the 1st BGRS Conference, Novosibirsk, ICG, SD RAS
- Ponomarenko J., Classification of eukaryotic transcription factors based on significant B-DNA conformational and phisical chemocal properties of their binding sites., Proc. of the 1st BGRS Conference, Novosibirsk, ICG, SD RAS, this issue.
- Suzuki M., Amano N., Kakinuma J., Tateno M. (1997) Use of a 3D structure data base for understanding sequence-dependent conformational aspects of DNA. J. Mol. Biol., 274, 421-435
- Fishburn P.C., Utility theory for decision making, NY, John Wiley & Sons (1970)
- Frolov A.S., et al., A system for activation of the TRRD database: further development of GeneExpress, Proc. of the 1st BGRS Conference, Novosibirsk, ICG, SD RAS, this issue, 9, 823-826.

Figure 1. Principle scheme of the GeneExpress system.

Figure 2. An example of the knowledge record: the C/EBP-transcription site consensus in the trinucleotide alphabet {MMM,MMK,MKM,MKK,KMM,KMK,KKM,KKK}.
 |
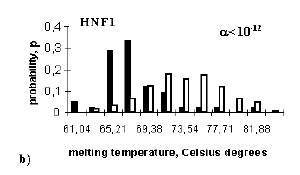 |
| Figure 3. Histograms of the mean value for transcription factor binding sites (black columns) and the random sequences (white columns): a) Ğroll angle in protein-DNA complexesğ averaged for the region [-10;2] for EN; b) the mean value of the melting temperature averaged for the region [-21;4] for HNF1. | |
 |
 |
 |
| Figure 4. The dependences of specific site activities on significant conformational and physical and chemical features: a) the transcription activity in vivo of PE1B region of the mouse a A-cristalline gene promoter (Sax et al., 1995) is determined primarily by probability DNA to be contacting with nucleosome core; b) the USF/DNA affinity (Bendall and Molloy, 1994) is determined by the B-helical twist angle; c) the CRO/DNA affinity (Kim JG et al., 1987) is determined by the major groove width. | ||