State Research Center of Virology and Biotechnology “Vector”, Koltsovo, Novosibirsk Region, Russia, 633159, e.mail: belova@vector.nsk.su
Keywords: interferon-induced Mx1 protein, mechanisms of expression regulation, gene net, model
1. Introduction
Interferons (IFNs) play a crucial role in the early host’s defense against viral infections. All cellular responses to IFNs result from interaction with cell surface receptors that trigger the expression of a number of cellular IFN-stimulated genes (ISGs). The system of regulation of gene expression, controlling Mx protein synthesis, is one of the key elements of IFN-induced antiviral resistance. Products of IFN-stimulated Mx genes represent the family of proteins with antiviral potential to influenza virus and various single-stranded RNA viruses [1-3]. The current theoretical study has been undertaken to elucidate mechanisms of Mx protein expression and antiviral action. The methods of mathematical modeling were used for analysis.
2. The regulation of Mx gene expression
Expression of Mx gene is mainly regulated by type I IFNs. The post-receptor JAK/STAT signaling pathway involves several steps, resulting in the protein phosphorylation and activation of transcription factors STAT1 (STAT ![]() ) and STAT2, formation of multimeric DNA-binding complex ISGF3 (IFN-stimulated gene factor) [4]. Complex ISGF3 in turn migrates to the nucleus and binds to specific DNA sequences known as IFN-stimulated response element (ISRE) upstream of Mx gene [5].
) and STAT2, formation of multimeric DNA-binding complex ISGF3 (IFN-stimulated gene factor) [4]. Complex ISGF3 in turn migrates to the nucleus and binds to specific DNA sequences known as IFN-stimulated response element (ISRE) upstream of Mx gene [5].
But virus infection or administration of double-stranded RNA (dsRNA) per se can also produce a quick and efficient Mx gene activation [5, 6]. In all cases Mx induction is a true primary response to the virus, rather than a secondary response to virus-induced IFN [5, 6]. Cells are capable of reacting rapidly on infection by simultaneously synthesizing Mx protein that will remain intracellular and IFNs that will be released into the cellular environment. This interferon induces expression of Mx protein in neighboring uninfected cells, such that the cells initially infected soon become demarcated by a barrier of a virus-protected cells. Consequently, virus can not spread efficiently, giving the immune system enough time to mount its own line of defense and eliminate the virus.
Despite the significance of direct activation of Mx by viral infection, little has been characterized of this pathway at the molecular level. Data permit to propose that this way of Mx induction differs from Mx induction by IFN. The investigation of virus- and dsRNA-dependent induction of other IFN-stimulated genes gives some representations about possible mechanisms of transcriptional regulation of Mx genes. First, it is known, that a DNA sequence containing the ISRE can mediate a transcriptional response to virus [7, 8]. The data indicate that ISRE-mediated signal transduction by dsRNA is distinct from ISRE-mediated IFN-![]() signaling [4] and NF-kB-mediated signaling by dsRNA or virus infection [9]. Second, ISG induction by viral infection is independent of ISGF3 activation [8]. In contrast to the lack of a role of ISGF3, several transcription factors, IRF-1, IRF-3, DRAF-1 and DRAF-2, which participate in activation of a few ISGs by viruses and dsRNA, were identified [6, 8, 10].
signaling [4] and NF-kB-mediated signaling by dsRNA or virus infection [9]. Second, ISG induction by viral infection is independent of ISGF3 activation [8]. In contrast to the lack of a role of ISGF3, several transcription factors, IRF-1, IRF-3, DRAF-1 and DRAF-2, which participate in activation of a few ISGs by viruses and dsRNA, were identified [6, 8, 10].
The most probable mediator of Mx induction by virus is IRF-1 factor. The binding of virus-activated STAT1![]() with GAS element in IRF-1 promoter [6, 11] could lead to induction of IRF-1 gene expression and following ISRE-mediated Mx induction. A negative feedback in regulation of Mx gene transcription, connected with action of IRF-2 repressor [11], is assumed [6].
with GAS element in IRF-1 promoter [6, 11] could lead to induction of IRF-1 gene expression and following ISRE-mediated Mx induction. A negative feedback in regulation of Mx gene transcription, connected with action of IRF-2 repressor [11], is assumed [6].
It is known that Mx mRNA expression in response to virus and dsRNA is amplified considerably if cells were pretreated with IFN [12]. This suggests that virus may synergizise with IFN to increase the Mx expression. We assume that binding of IRF-1 and ISGF3 factors with ISRE sites in Mx1 promoter leads to synergistic transcriptional activation of Mx1.
More detailed description of these events and principles of gene net organization are presented in the data base IIG-TRRD [13] installed on WWW server on address: http://www.bionet.nsc.ru/trrd/.
3. Antiviral action of Mx1 protein
We have focused our attention on the next aspects of regulation of Mx1 system in mouse cells challenged with influenza A virus:
- Efficacy of cell protection provided by IFN- and virus-inducible Mx1.
- Efficacy of antiviral defense depends on the rate and the level of Mx1 induction. Role of synergistic transcriptional control of Mx1 level.
- Functional role of Mx1 oligomerization.
Mathematical model of regulation of Mx1 protein induction and action
Our first aim was to integrate particular in vivo and in vitro data sets including heterogeneous and incomplete observations into a conceptual model of regulation Mx1 protein induction and action in the course of influenza infection., with interactions described in terms of positive and negative influences. Both the course and the outcome of infection are influenced on the one hand by virus cytopathogenicity, rapidity of spread, generation time, susceptibility to interferon etc, and on the other hand by the efficiency of antiviral defense. There is a need to assess these factors in quantitative terms to provide a firm basis for further rigorous analysis of cell resistance to virus infection. The complexity of the system is so high that predictive understanding of the mechanisms of the antiviral action of Mx1 can not be achieved without careful mathematical modeling. The conceptual model was interpreted and organized into a mathematical model.
The variables of model are concentrations, respectively: V(t) – infective virus particles; I(t) – interferon; MI(t)- IFN-inducible Mx1 protein; MV(t) – virus-inducible Mx1 protein; C(t) – virus permissible (sensitive) cells; CI(t)- protected by IFN from viral infection (resistant) cells.
The model we propose consists of a set of six delay-differential equations.
 |
(1) |
| (2) | |
| (3) | |
| (4) | |
| (5) | |
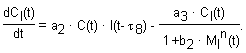 |
(6) |
| Initial conditions: |
(7) |
Physical meaning of parameters: a1 – the rate constant of virus binding with cells; a2 – the rate constant of IFN binding with intact cells and of induction of cell’s virus resistant state; characterizes the kinetic of intact cell transition to the virus resistant state, directed by intercellular IFN; a3 – the rate constant of decay of cell’s virus resistant state; characterizes the process of transition of the protected cells to the virus permissible state, in the absence of intercellular IFN; d – virus yield (virus per cell); q1, q2 – the numbers of receptors to virus and IFN per cell; p1, p2 – the rates of IFN production by sensitive and resistant cells; m1, m2 – the rates of virus-inducible Mx1 production by sensitive and resistant (IFN- pretreated) cells; m3 – the rate constant of IFN-inducible Mx1 production by sensitive cells; k1, k2, k3 – the rate constants of nonspecific virus removal and decay of IFN and Mx1; b1 – the rate constant of inhibition of virus reproduction by virus-induced Mx1; b2 – the rate constant of inhibition of decay of virus resistant state of cells by IFN-induced Mx1; n – the number of Mx1 proteins in a oligomer structure; C* – the concentration of cells in an initial moment of time. The duration of processes is taken into account by delays.
5. Results and discussion
For identification of model structure and parameters we used the experimental data in vitro, describing kinetics of main variables of model [14]. The results obtained by numerical solution of the model equations provide a good agreement with the data observed in experiments. The model reproduces the main behavior regularities of system “virus-interferon-Mx1” under varying of multiplicity of infection, initial interferon concentration, and genotype of cell culture (Mx1-positive or -negative). Our results allow to evaluate the role of mechanisms of virus and IFN-mediated Mx1 expression in induction of antiviral state.
A subtle balance exists between the infecting virus and the mobilization of early defense forces. It is conceivable that the localization and timely mobilization of these defense forces is very important. The regularities of influenza virus multiplication and different production strategies of virus- and IFN-inducible Mx1 proteins permit to assume that Mx1 proteins being induced by means of alternative mechanisms inhibit different steps of the virus multiplication cycle and this results in different efficacy of cell protection. As was shown by computer analysis, virus-induced Mx1 can not provide protection of IFN-untreated cells challenged with high or low doses. In this case, Mx1 protein produces in infected cells in late stage of virus replication cycle and therefore is unable to block virus spread. Endogenous IFN induces in reply on virus late enough and also is unable to protect cells timely. On the contrary, exogenous or spontaneously releasing IFN, appearing in culture prior to infection, induces early Mx1 production and protects cells under any virus dose.
The most important is a rapid expression of the antiviral protein Mx1 at precisely the sites where the virus initially replicates. According to simulation results, the level of Mx1 protein required to block virus spread was achieved only under synergistic activation of Mx1 expression by virus and IFN in cells pretreated with IFN. The number of cells in which Mx1 is induced also appears to be one of the major factors of antiviral resistance. Really, model exploration demonstrated that one of the reason of destruction of the IFN-pretreated Mx1-negative cells is the lower part of protected cells in this culture compared with the part of such cells in culture of Mx1-positive ones. Oligomerization of Mx1 protein can be the factor, which provides the most adequate antiviral response.
It seems important to explore the point that the model developed provides a tool for knowledge organization by relating different particular factors and heterogeneous data into an integrated and consistent quantitative description of infection that gives a basis for further analysis. The received results give a new knowledge about the key aspects of regulation of influenza virus-IFN-Mx1 system.
References
- Horisberger, M.A. (1995). Am. J. Respir. Crit. Care Med. 152, 567-571.
- Arnheiter, H., Frese, M., Kambadur, R., Meier, E. & Haller, O. (1996). Curr. Top. Microbiol. Immunol. 206, 119-147.
- Frese, M., Kochs, G., Feldmann, H., Hertkorn, C. & Haller, O. (1996). J. Virol. 70, 915-923.
- Levy, D. (1995). Sem. Virol. 6, 181-189.
- Hug H., Costas M., Staeheli, P., Aebi, M. & Weissmann, C. (1988). Mol. Cell. Biol. 8, 3065-3079.
- Ronni, T., Sareneva, T., Pirhonen, J. & Julkunen, I. (1995). J. Immunol. 154, 2764-2774.
- Daly, Ch. & Reich, N.C. (1995). J. Biol. Chem. 270, 23739-23746.
- Bandyopadhyay, S.K., Leonard, G.T., Jr., Bandyopadhyay, T., Stark, G.R. & Sen, G.C. (1995). J. Biol. Chem. 270, 19624-19629.
- Hiscott, J., Nguyen, H. & Lin, R. (1995). Sem. Virol. 6, 161-173.
- Grant, C.E., Vasa, M. Z. & Deeley, R.G. (1995). Nucl. Acids Res. 23, 2137-2146.
- Pine, R., Canova, A. & Schindler, C. (1994). EMBO J. 13, 158-165.
- Goetschy, J.-F., Zeller, H., Content, J. & Horisberger, M.A. (1989). J. Virol. 63, 2616-2622.
- Ananko, E.A., Bazhan, S.I., Belova, O.E. & Kel A.E. (1997). Molec. Biology, 31, 701-713.
- Arnheiter, H., Haller, O. & Lindenmann, J. (1980). Virology 103, 11-20.