KEL A.E.+, KEL-MARGOULIS O.V., BABENKO V.N., WINGENDER E.#
Institute of Cytology and Genetics, (Siberian Branch of the Russian Academy of Sciences), 10 Lavrentieva ave., Novosibirsk, 630090 Russia, kel@bionet.nsc.ru;
#Gesellschaft fur Biotechnologische Forschung mbH, Mascheroder Weg 1, D-38124 Braunschweig, Germany, ewi@gbf.de
+Corresponding author
Keywords: promoters, immune cell activation, eukariotic organisms, regulatory regions, transcription factor binding sites, weight matrix
We have developed a new method for computer prediction of gene expression profile by identifying specific regulatory sequences in promoters and in other transcription regulatory regions. The method is based on recognition of highly specific composite elements (CE) – complex regulatory units consisting of synergistically functioning transcription factor binding sites, providing the first level of combinatorial transcriptional regulation. The method has been applied to the identification of genes induced upon immune cell activation. Immune cell specific composite elements were modelled. The NFAT/AP-1 model was tested for recognition of potential CEs of this type in a large set of immune cell specific genes. The frequency of potential composite elements within promoters and enhancers of genes which are expressed during T-cell activation is 3-times higher than within the coding regions of these genes and 4-times higher than in muscle-specific promoters.
1. Introduction
Combinatorial regulation is a basic mechanism of gene expression control in eukaryotic organisms [1]. The pattern of expression of eukaryotic genes is encoded mainly in the structure of their transcription regulatory regions by arrays of transcription factor (TF) binding sites. These sites are organised in a hierarchical manner, constituting several levels of combinatorial complexity. The lowest level of this combinatorial regulation is provided by composite regulatory elements – combination of target sites for two different transcription factors, that interact with each other resulting in a particular expression pattern of genes containing this CE.
The rapid accumulation of sequence data faces molecular biologists with a new challenge to identify and functionally characterize all genes expressed in living organisms. Transcription control of gene expression is the clue to understand the functional role of genes in maintenance and development of higher eukaryotic organisms. Urgently needed are computational approaches by which we could check hypotheses about possible features of transcriptional regulation of genes from sequence data. There are several attempts in creating computational approaches for utilizing information on combinatory of transcription factor binding sites for analysis and even classification of eukaryotic promoters and other transcription regulatory regions [2,3,4].
There is a large number of genes whose expression is restricted to T- and B-cells and is dramatically increased after antigenic stimulation. A number of different transcription factors is involved in the combinatorial regulation of genes expressed in the activated immune cells. Some of these genes have been shown to contain closely situated binding sites for NFAT and AP-1 within their regulatory regions. Co-operation between NFAT and AP-1 provides a mechanism for combinatorial transcriptional regulation, which integrates calcium-dependent and protein kinase C-dependent pathways of immune-cell activation.
Our ultimate goal is to reveal all requirements on structural organization of transcription regulatory regions of the genes involved in immune response that provide formation of different transcription complexes consisting of many transcription factors giving rise to the combinatorial regulation of expression of these genes. As the first step towards this goal we try to model the NFAT/AP-1 composite elements in the regulatory regions of genes induced upon immune cell activation.
2. Data and Methods
2.1 Set of experimentally proven NFAT/AP-1 composite elements
The set of 13 NFAT/AP-1 CEs was extracted from the COMPEL database release 2.1 [5,6]. Some features are common to all of them. First, two binding sites are immediately adjacent or even overlap, and the distance between them varies between 5 to 11 nucleotides (distance between the second G in the GGAAA core of NFAT site and the central nucleotide in the TGASTCA core of AP-1 site). This characteristic of CEs is referred to as a „distance rule“. Second, the AP-1 site is always located at the 3′ side of the GGAAA consensus of the NFAT site. We will refer to this feature as „orientation rule“. And third, the binding affinity of these two factors varies coordinately among composite elements. For example, binding of NFAT to its high affinity site was shown to stabilize interaction of AP-1 with an adjacent low affinity site [7].
2.2 Sets of sequences used for recognition of potential composite elements
There were two main sequence sets where we searched for potential composite elements. Set T includes known and potential target genes for NFAT/AP-1 regulation. Set M contains sequences for muscle-specific genes. In set T, we collected EMBL sequences of genes that are known to contain NFAT/AP-1 CEs or, at least, to be expressed in activated T- , B- and/or mast cells and the activation of which is sensitive to the immunosuppressant cyclosporin A. The T-set contains cytokine genes, genes for some surface receptors and some specific transcription factors of different mammalian species. The M-set contains genes whose expression is confined to muscle cells, including contractile proteins and certain transcription factors. Since the expression pattern of these genes is quite different from that of T- and B-cell genes, we have chosen M as a control set for our analysis.
2.3 Position Weight Matrix (PWM) method for recognition of individual sites
A modification of PWM method with information vector transformation [8] was used for recognition of individual NFAT and AP-1 sites in the sequences. The following score q is calculated for a sliding window of length w in a sequence under study.
| (1) | |
| with information vector: |
(2) |
here, ![]() stands for the frequency of the nucleotide b (
stands for the frequency of the nucleotide b (![]() ) in position i of the PWM.
) in position i of the PWM. ![]() – are minimal and maximal frequencies in the i-th position of the PWM. We have shown that an estimation of free energy E of transcription factor binding to the target sites could be done using the score q:
– are minimal and maximal frequencies in the i-th position of the PWM. We have shown that an estimation of free energy E of transcription factor binding to the target sites could be done using the score q: ![]() .
.
The weight matrixes for NFAT and AP-1 sites were created on the basis of corresponding sets of sites collected in the TRANSFAC 3.3 and TRRD 3.5 [6] databases. The set of AP-1 sites includes 47 sequences. The set of NFAT binding sites comprises 41 sequences.
2.4 Constructing the method for recognition of NFAT/AP-1 CEs.
For each CE, two parameters: ![]() and
and ![]() are calculated for the two corresponding binding sites constituting the composite element. It is known that in composite elements one of the factors may stabilize the interaction of the second factor with a comparatively poor binding site. To model such a phenomenon we constructed a method for recognition of composite elements by combining two parameters:
are calculated for the two corresponding binding sites constituting the composite element. It is known that in composite elements one of the factors may stabilize the interaction of the second factor with a comparatively poor binding site. To model such a phenomenon we constructed a method for recognition of composite elements by combining two parameters: ![]() and
and ![]() . For combining these two parameters into one recognition function we use the SITEVIDEO software [9] which provides means for obtaining the best discrimination between training set of CEs and control data (random sequences). The resulting recognition function which we call composite score qCE is the following.
. For combining these two parameters into one recognition function we use the SITEVIDEO software [9] which provides means for obtaining the best discrimination between training set of CEs and control data (random sequences). The resulting recognition function which we call composite score qCE is the following.
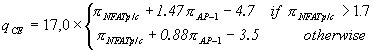 |
(3) |
3. Results and Discussion
3.1 NFAT/AP-1 CEs are enriched within promoters of genes induced upon immune cell activation.
In order to investigate whether there is a function-correlated enrichment of NFAT/AP-1 CEs in genomic sequences we performed extensive search for potential CEs in promoters, exons and introns of T- and M- sets. We did the search with various threshold levels for the CEs and individual sites.
Totally, sequences of T-set are 1.7 times more enriched by potential NFAT/AP-1 CEs than sequences of M-set. Moreover, when we analysed different functional parts of the genes separately we gained an even more profound difference between sequences from T- and M-sets. It turned out that promoters of T-set genes reveal the higher concentration of NFAT/AP-1 CEs. They contain potential CEs up to 4-times more frequently than muscle-specific promoters (see Fig. 1). Interestingly, the difference increases while making the search mode more restrictive. So, the high scoring CEs are the most specific for immune-cell specific gene promoters and could be used as a benchmarks for this type of genes.
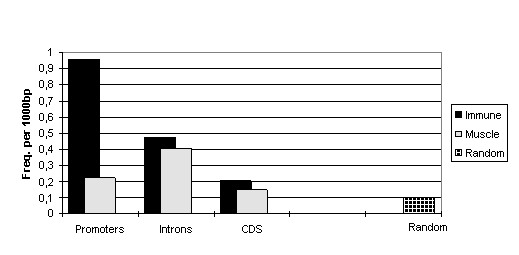
Figure 1. Frequencies of NFAT/AP-1 composite elements (qCE >10.0) in the functional parts of immune-cell specific genes, muscle-specific genes and random sequences.
3.2 Location of NFAT/AP-1 CEs near transcription start is a specific feature of genes induced upon T-cell activation.
To understand a role of NFAT/AP-1 CEs in transcription activation of T-cell specific genes we analysed the location of potential CEs found in different promoter sequences. Promoters from T and M sets were divided into subregions of length 150bp relative to transcription start, and frequencies of potential CEs in these subregions were analysed. In the Figure 2 we present a bar diagram with relative frequencies of the CE in different promoter subregions. One can see that the region from -300 to the start of transcription of T-promoters is the most enriched in CEs. In contrast, very low frequency of the CEs in the same region is a characteristic feature of M-set promoters. In close proximity to transcription start (up to -150bp) the frequencies differ 10-times and more.
Thus, we can clearly classify promoters to T- or M- groups based on frequencies of potential CEs and on the presence of potential binding sites within the first 300 bp upstream of transcription start site. This classification rule has a high recognition potential: 10% to 18% of false negatives with less then 10% of false positives.

Figure 2 Bar diagram of relative frequencies of NFAT/AP-1 CEs (qCE>0.0) in different subregions upstream start of transcription.
3.3 Screening of EMBL nucleotide sequence database for potential NFAT/AP-1 CEs.
We performed a search of potential CEs through EMBL database sections containing genes of mammalian organisms (sections: hum1, hum2, rod, mam). The task was to check promoter regions of the genes and to identify those that contain NFAT/AP-1 CEs. In the course of the screening we applied the “300bp rule”. The rule requires that at least one potential CE should be found in close proximity (less then 300bp) to the transcription start. Only promoters with CE frequency ![]() >1.7 per 1000bp were selected. In addition, we performed a cluster analysis of the found CEs. We require that at least one cluster of 4-5 CEs should be found in the upstream promoter sequence. In total 2304 promoters of mammalian genes were scanned. 81 promoters passed through the selection rules and were picked up by the program. 27 (33%) promoters were for inflammatory cytokine genes and their receptors; 14 (17%) promoters were for other relevant genes, such as genes involved in immune and acute-phase response (serum amyloid genes, genes for immunoglobulins and MHC complex, genes involved in regulation of immune cells proliferation) as well as genes involved in intracellular signaling during T- and B-cell activation. In addition 28 (35%) promoters of probably irrelevant genes were selected by the program and 12 (15%) promoters of genes with unknown function (EMBL annotation of these genes was done in long anonymous genomic sequences on the basis of multiple homology with cDNA fragments).
>1.7 per 1000bp were selected. In addition, we performed a cluster analysis of the found CEs. We require that at least one cluster of 4-5 CEs should be found in the upstream promoter sequence. In total 2304 promoters of mammalian genes were scanned. 81 promoters passed through the selection rules and were picked up by the program. 27 (33%) promoters were for inflammatory cytokine genes and their receptors; 14 (17%) promoters were for other relevant genes, such as genes involved in immune and acute-phase response (serum amyloid genes, genes for immunoglobulins and MHC complex, genes involved in regulation of immune cells proliferation) as well as genes involved in intracellular signaling during T- and B-cell activation. In addition 28 (35%) promoters of probably irrelevant genes were selected by the program and 12 (15%) promoters of genes with unknown function (EMBL annotation of these genes was done in long anonymous genomic sequences on the basis of multiple homology with cDNA fragments).
Acknowledgments
Different parts of this work were funded by the European Commission (BIO4 CT950226), by the Bundesministerium fur Bildung, Wissenschaft, Forschung und Technologie (project no. X224.6), by the Russian Ministry of Sciences and the Russian Fundamental Research Foundation (grants: 97-04-49740, 96-04-50006), by the Siberian Branch of Russian Academy of Sciences, as well as by the North Atlantic Treaty Organization (grant no. 951149).
References
- P.Ernst and S.T.Smale “Combinatorial regulation of transcription I: general aspects of transcriptional control” Immunity 2, 311-319 (1995)
- Y.V.Kondrakhin , A.E.Kel , N.A.Kolchanov, A.G.Romashchenko, L.Milanesi “Eukaryotic promoter recognition by binding sites for transcription factors” Comput.Applic.Biosci. 11, 477-488 (1995)
- J.W.Fickett “Quantitative discrimination of MEF2 sites” Mol.Cell.Biol 16, 437-441 (1996)
- K. Frech, K. Quandt and T. Werner “Muscle actin genes: A first step towards computational classification of tissue specific promoters.” In Silico Biol. 1, 0005. <http://www.bioinfo.de/isb/1998/01/0005/> (1998)
- O.V. Kel, A.G. Romaschenko, A.E. Kel, E. Wingender and N.A. Kolchanov “A compilation of composite regulatory elements affecting gene transcription in vertebrates” Nucleic Acids Res. 23, 4097-4103 (1995)
- T. Heinemeyer, E. Wingender, I. Reuter, H. Hermjakob, A. E. Kel, O. V. Kel, E.V. Ignatieva, E.A. Ananko, O.A. Podkolodnaya, F. A. Kolpakov, N. L. Podkolodny and N. A. Kolchanov “Databases on transcription regulation: TRANSFAC,TRRD and COMPEL” Nucleic Acids Res. 26, 362-367, (1998)
- J. Jain, P.G. McCaffrey, Z. Miner, T.K. Kerppola, J.N. Lambert, G.L. Verdine, T. Curran, and A. Rao “The T-cell transcription factor NFATp is substrate for calcineurin and interacts with Fos and Jun” Nature 365, 352-355 (1993)
- K. Quandt, K. Frech, H. Karas, E. Wingender, and T. Werner “MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data” Nucleic Acids Res. 23, 4878-4884 (1995)
- A.E. Kel, M.P. Ponomarenko, E. Likhachev, Yu.L. Orlov, I.V. Ischenko, L. Milanesi, N.A. Kolchanov “SITEVIDEO: a computer system for functional site analysis and recognition. Investigation of the human splice sites” Comput.Applic.Biosci. 9, 617-627 (1993)