BLINOV V.M.+, RESENCHUK S.M., CHIRIKOVA G.B., PETROV N.A., PUZYREV A.A.1, ORESHKOVA S.F.1, KUVSHINOV V.N.1, ILYICHEV A.A.1, ROMASHCHENKO A.G.2, RUZANKINA Ya.S.2, MARTYNOV Yu.A.2, VINOGRADOV S.S.2, VOEVODA M.I.2, VARDASANIDZE K.S.4, KISSELEV L.L.5, MARTYNYUK R.A., SANDAKHCHIEV L.S., KARAKIN E.I.3
Institute of Molecular Biology, SRC VB “Vector”, Koltsovo, Novosibirsk region, 633159, Russia; e-mail: blinov@vector.nsk.su;
1Institute of Biotechnology and Bioengineering, SRC VB “Vector”, Koltsovo, Novosibirsk region, 633159, Russia;
2Interinstitutional Laboratory of Human Molecular Epidemiology and Evolution, Institute of Cytology and Genetics, SB RAS, and Institute of Internal Medicine, SB RAMS, 10 Lavrentiev Ave., Novosibirsk, 630090, Russia;
3Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, 10 Lavrentiev Ave., Novosibirsk, 630090, Russia;
4Novosibirsk State Medical Institute, Novosibirsk, Russia;
5Engelhardt Institute of Molecular Biology, RAS, ul. Vavilova, 32, 117984 Moscow, Russia;
+Corresponding author
Keywords: P53, hypersensitive sites, mRNA forms, DNA/RNA secondary structure, exons, adduct formations, benzopyrene
Analysis of the DNA isolated form biopsies of cancer patients allowed us to map single and double mutations at codons 242, 245, and 248 in tumor-suppressing gene p53. The invariant DNA/RNA secondary structures for exons 5-8 of p53 gene were calculated. The models of aflatoxin B1 and benzopyrene interactions with the critical codons 249/248 were created: these codons were shown to be located in the looped regions. In the paired regions, we have located compensatory substitutions that can have a selective advantage compared with single substitutions, change discretely the p53 function and may have a substrate specificity. Statistical analysis of the mutations and comparison of the invariant RNA/DNA secondary structures of the normal and mutant forms of p53 allowed us to reveal the so-called “superhot oncogenic” mutations. In addition, such invariant regions of p53 gene displaying high frequency of paired mutations are capable of forming potential homo- and heteroduplexes and, therefore, are potential recombination sites. Mechanisms of p53 gene function and balance between the normal and mutant p53 forms are discussed.
1. Introduction
Gene p53 occupies a special position among the marker genes for cancer diagnostics: it is mutated in approximately 50% tumors. The majority of point mutations in this gene (90%) are sense mutations causing changes in p53 function; the rest mutations are connected with deletions, insertions, and damages at splicing sites and cause premature translation termination and complete inactivation of p53. Distributions and number of the most frequently occurring mutations at different codons depends both on the type of tumor and exogenous factors (carcinogens and mutagens) capable of adduct formation with “hotspots ” of p53 gene The majority of p53 gene mutations in human tumor cells either alter or destroy completely the transcriptional activity of p53. The loss of p53 function can cause an uncontrolled cell proliferation. A considerable increase in the level of p53 is a typical organismal response to environmental stress effects [1]. All these allows p53 to be described as a “guardian of the genome’s integrity”.
Inactivation of tumor-suppressing p53 gene through single point mutations transfers a normal cell into a tumor cell. Certain mutations not only inactivate the suppressor function of p53 but result simultaneously in dominant oncogenic properties of p53; thereby transforming the antioncogene p53(+) into the oncogene p53(-). The role of mutant p53(-) in regulation of the genes involved in cell proliferation and tumor formation is yet vague. For instance, mutant p53(-) variants were shown to regulate the expression of mdr-1 gene in NIH3T3 cells [2], to activate vascular endothelial growth factor in embryonic kidney cells, to stimulate the expression of proliferating cell nuclear antigen [3], to be involved in formation of cross-like looped single-stranded DNA regions aiding to the activation of chromatin through specialized MAR/SAR recognition sites. Fixing of DNA loops is likely to depend on the different states of cells connected with the state of chromatin: active gene expression, replication, or differentiation.
Highly mutagenic deamination of cytosine and 5-methylcytosine is most frequent in single-stranded DNA regions. Initiation of meiotic recombination occurs preferentially in the DNA regions located in “open” chromatin conformations, which are, thus, capable of forming heteroduplexes between single-stranded regions belonging to different chromosomes. Hence, it is supposed that the recombination takes place in those chromosome regions where the greatest number of mutations occur; and vice versa, mutations occur preferentially in the recombination sites.
Mutations in p53 gene cause different tumor types in human organism, and most of these mutations fall within the central domain of p53 gene. Virtually all these mutations change the function of p53 and drastically affect the stability of p53 protein. Mutant forms can display abnormal conformations and the lifetime increased to several hours, whereas the normal lifetime is 20 min. The central domain of p53 (102-292) recognizes specifically four consensus repeats (5′-PuPuPuC(A/T)-3′) [4]; amino acid residues Lys-120, Cys-277, and Arg-280 form hydrogen bonds with nucleotide bases., and the residues Ser-241, Arg-273, Ala-276, Arg-283, and Arg-248 interact with the phosphate groups in the major and minor groves of the consensus repeat.
2. Results and discussion
Analysis of the DNA isolated from tumor biopsies of cancer patients allowed us to map single and double mutations at codons 242, 245, and 248 in the tumor-suppressing gene p53 (Fig. 1) [5].

Fig. 1. The nucleotide sequence of exon 7 of p53 gene [6]. The point mutations found in the DNA isolated from biopsies of oncological patients 1T7A, 4T7D, 4T7A, 4T7C and the corresponding amino acid substitutions in mutant p53 proteins are indicated. Direct and inverted repeats (CCATC, ACTACA, GCATG, and ACTGT) are shown.

Fig. 2. (A) The model of adduct formation between aflatoxin B1 and codon 249 (AGG; Arg) located in a looped single-stranded DNA (or mRNA) region within exon 7 of p53 gene. The interaction energy of the paired regions in the invariant DNA/RNA structure in exon 7 amounts to –21.3 kcal/mol; complementary nucleotides are indicated with (*).
(B) The model of adduct formation between aflatoxin B1 and codon 249 (AGG; Arg) within a heteroduplex between two homologous DNA regions in exons 7 of p53 genes. The interaction energy of the paired regions amounts to –48.6 kcal/mol; complementary nucleotides are indicated with (*).
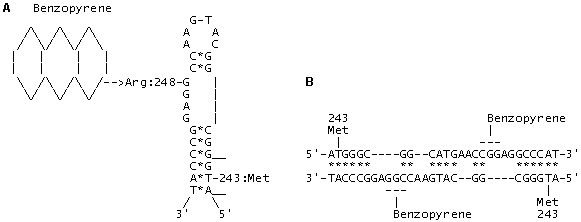
Fig. 3. (A) The model of adduct formation between benzopyrene and codon 248 (CGG; Arg) located in a looped single-stranded DNA (or mRNA) region within exon 7 of p53 gene. The interaction energy of the paired regions in the alternative invariant secondary DNA/RNA structure in exon 7 amounts to –22.6 kcal/mol; complementary nucleotides are indicated with (*).
(B) The model of adduct formation between benzopyrene and codon 248 (CGG; Arg) within a heteroduplex between two homologous DNA regions in exons 7 of p53 genes. The interaction energy of the paired regions in heteroduplex amounts to –51.2 kcal/mol; complementary nucleotides are indicated with (*).
The invariant DNA/RNA secondary structures for exons 5-8 of p53 gene were calculated basing on the epidemiological data on the mutations in p53 and the revealed “hotspots”—targets of forming the mutagen and carcinogen adducts with DNA. The models of aflatoxin B1 and benzopyrene with the critical codons 249/248 were created: these codons were shown to be located in the looped regions (Figs. 2 and 3). In the paired regions, we have located compensatory substitutions that can have a selective advantage compared with single substitutions, change discretely the p53 function and may have a substrate specificity. Statistical analysis of the mutations and comparison of the invariant RNA/DNA secondary structures of the normal and mutant forms of p53 allowed us to reveal the so-called “superhot oncogenic” mutations. In addition, such invariant regions of p53 gene displaying high frequency of paired mutations are capable of forming potential homo- and heteroduplexes and, therefore, are potential recombination sites. Origination of such induced paired mutations according to the principle of compensation in primary and metastatic tumors is to be clarified along with the formulation of the necessary and sufficient conditions for transition of antioncogene p53(+) to oncogene p53 (-). The number of mutations in exons 5, 7, and 8 is 4408, that is, 80% of all the mutations found in these tumors, while the number of paired compensatory mutations at codons 245-248, 175-181/183, and 273-282/283 in p53 amounts to 1368 (30%). The same codons were recently fount to house the mutations imparting resistance to chemotherapeutic preparations of doxorubicin, cisplatin, and taxol types.
Double selection of p53 DNA regions and possible binding of the protein to its own hotspots can lead to self-regulation of the gene itself. The mutations are clustered preferentially in permitted sites that are variable and conservative structures simultaneously. Protein p53 can partially unwind the DNA regions housing the recognition half-site [5′-PuPuPuC(A/T)-3′] and hotspots through interaction with exons 5 and 7. Such tuning and “self-damaging” only in the permitted compensatory codons means that the function of p53 is changing in response to the environmental conditions. This “mutation” mechanism is very similar to editing typical of a number of viral genomes [7] and certain human genes. As a result, one gene can code for several functions depending on the damage extent of the cell genetic machinery and switch on alternative ways of salvation of the entire organism at the expense of either programmed local cell death, or their transformation into tumor cells. And p53 plays the key role in these processes.
Does the balance between p53(+) and p53 (-) exist, such a universal evolutionary pathway of p53 implying the coexistence of normal p53(+) forms and a limited number of discrete mutant p53(-) forms provided? Is the transition from p53(-) to p53(+) possible and can it be connected with fixation of compensatory mutations, as a result of which the mutant p53 becomes nonmutant and able to recognize the binding site correctly? The trigger mechanism of p53 action that includes the feedback must constantly improve the self-regulation system on the whole. Permitted compensatory mutations generate adaptive alternative forms of p53 protein, in which the amino acids involved in the interaction with DNA are selectively advantageous.
Connections of the codons in conformations R(249-) and R(249+) are different: a change in the codon 243R(249-) induces the changes in codons 253 and 254, whereas a change in the codon 243R(249+) induces the changes in codons 250 and 251. It is these coupled and permitted interaction chains that, on the one hand, predetermine the discrete conformations of p53 protein and, on the other, cause the asymmetric distribution of double compensatory mutations among other “metastable” mutations. The self-editing of p53 takes place depending on stress conditions. The number of such states is stringently fixed and determined by the invariant secondary structures of exons 5, 7, and 8. It is these invariant structures that interact with the external factors, carcinogens, and represent the major internal sites for transcription factor p53 simultaneously. We don’t know the complete list of cellular and viral genes and their products that are involved into protein-protein interactions with p53. This is also true for viral and cellular genes containing half-sites for p53 activation/inhibition, which are involved in the protein-nucleic interactions with p53. We also know nothing about the limitations to carcinogens and other external stress factors that are capable of adduct forming with p53 gene regions. The invariant structures containing the compensatory mutations in p53 that we revealed is likely to help in answering a number of the above-mentioned questions.
What is the volume of “point mutation load” that p53 can bear and what are the limits within which the compensatory mechanism of mutation origination can provide for the two basic control functions of this gene? The protection system of the cell and organism on the whole is provided for by the system of control gene p53; therefore, any damage within this gene leads to extraordinary events. That is why the safety of the p53 system and its correct programmed function is likely to prevent any unexpected situations. Since the preservation of the p53 function as a “guardian of the genome’s integrity” requires switch-on of reparation, transcription, and recombination systems, the two potential recombinogenic regions in the exons 5 and 7, which we discovered, may play a most important role. The crossover at these sites between mutant and normal copies of p53 gene can revert the normal phenotype p53(+) and discard the accumulated mutation load. How these endogenous recombination events of reverting the p53(-) to p53(+) phenotype can be activated? If the p53 system is a “Ministry of Emergency Situations” responding to internal and external stresses, why it allows to “self-damage and self-edit” itself only in a limited number of hotspots, which are simultaneously the recombination sites and sites for interaction with carcinogens? The questions are yet to be answered.
Acknowledgments
The work was supported by the Russian Ministry of Science, Moscow Government, and Mayor of Moscow Yu.M. Luzhkov within the Program “Development and promotion of new methods for diagnostics and treatment of oncological diseases. The authors are grateful to Neal Cariello and Thierry Soussi for the database on p53 mutations and to the Russian National Program Human Genome for CD-based genomic databases.
References
- L.S. Cox and D.P. Lane, BioEssay, 17, 501 (1995).
- R.L. Zastawny, et al., Oncogene, 8, 1529 (1993).
- B.F. Muller, et al., Oncogene, 12, 1941 (1996).
- Y. Cho, et al., Science,. 265, 346 (1994).
- V.M. Blinov, et al., Mol. Biol. (Mosk.)(1998, in press).
- V.L. Buchman, et al., Gene, 70, 245 (1988).
- A. Sanchez, et al., Proc. Natl. Acad. Sci. USA, 93, 3602 (1996).