MILANESI L.1+, ROGOZIN I.B.2, D’ANGELO D.1
1Institute of Advanced Biomedical Technologies ITBA-CBR, Via Fratelli Cervi, 93, 20090 Segrate (Milan), Italy;
e-mail: milanesi@itba.mi.cnr.it;
2Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences 10 Lavrentiev Ave., Novosibisk 630090, Russia;
+Corresponding author
Keywords: protein coding genes, prediction tools, splice sites, homology search, integrated coding potential, TATA-box signals, polyadenilation signals, binding sites, repeated elements
1. Introduction
The WebGene suite of programs have been created to predict gene structure models in different organisms taking into account coding regions, functional signals and other properties in genomic sequences [1-4]. In particular the WebGene system is able to perform the following procedures:
- Identification of the potential splice sites, start and stop codons.
- Homology search with the EST database.
- Estimation of an integrated coding potential of the revealed PCF by using dicodon statistics and homology with key proteins and ESTs.
- Construction of a potential gene models from the set of potential coding regions, potential splice sites, start and stop codons.
- TATA-box signals prediction.
- Polyadenilation signals prediction.
- Binding sites prediction of transcription factors.
- Automatic homologies searching in protein database for analysis of the predicted protein.
- Identification of CpG islands.
- Identification and mask for repeated elements in a query sequence.
- Retrieval and visualization of the most relevant informations from a set of biological databases.
2. Description
The programs and modules implemented in the WebGene system can facilitate an iterative analysis of a new DNA sequence. A short descriptions of the most relevant options of the programs used in the WebGene system are:
2.1. Mode of splice sites prediction
Splice sites are predicted by combining classification approach and weight matrix.
Best splice sites – about 5% of real splice sites will be lost, but overprediction is small (about 15% of pseudosites will be predicted as splicing signals).
Almost all real splice sites – splice sites are predicted by using weight matrix technique. As result, about 2% of real splice sites will be lost, but a number of false sites will be predicted (30-35%).
2.2. Potential coding regions prediction
All potential exons – almost all potential coding exons (with and without similarity to key proteins) will be used for gene reconstruction.
Good exons – exons with good coding potential (with and without similarity to key proteins) will be used for gene reconstruction.
Excellent exons – only exons with excellent coding potential (with and without similarity to key proteins) will be used for gene reconstruction.
Only exons with similarity to key protein – only exons with marginal and significant similarity to key protein will be used for gene reconstruction. Some false exons can be included in prediction.
Only exons with expressed similarity – only exons with significant similarity to key protein will be used for gene reconstruction.
2.3. First and last coding exons
First and last exons with expressed similarity – this option puts limitations on the quality of the first and the last exons: gene will start and finish with well confirmed coding exons. This option is very important if several genes are located in a query sequence. No restriction on first and last exons – Any exons can be used as first and last ones.
2.4. Complete gene model
This option gives priority to models of complete (with the first and last exons) potential gene structure.
2.5. Sequence range for coding regions prediction
This option is very important in the case of several genes present in a query sequence. Default values are the first and the last positions of a query sequence.
2.6. Repeated elements detection
Prediction of repeated elements in newly sequenced DNA becomes very important in large genome sequencing projects, since presence of repeats in a query sequence can create a lot of problems for homology searches. This problem is complicated due to high heterogeneity and short length of repeated elements. Homology searches against the collection of repeated elements are used for repeats revealing. We have implemented a REPEAT program based on statistical estimates. Results of REPEAT are using in the ESTMAP and WebGene systems for ESTs mapping, although output information about repeats is much more brief in ESTMAP and WebGene. A masked sequence (with ‘N’s instead of repeated elements) is produced by the REPEAT program.
2.7. Homology search with the EST database
Currently existing collections of expressed sequence tags (ESTs) are very large and thus very useful for gene mapping. Homology searches against the EST Division of GenBank (dbEST) can be used for this purpose.
2.8. Database Homology search
Further analysis of predicted protein can help to confirm the gene prediction results, although such analysis is very difficult for proteins without significant homologies to known proteins already present in the databases. We implemented homology searches through SWISSPROT for analysis of the predicted protein.
2.9. CpG island prediction
CpG island can be very important for gene identification, since these are often considered to be gene marks and are frequently found at the 5’ ends of genes.
2.10. Organisms
WebGene system is capable to analysing the genomic sequence of: human, mouse, Drosophila, C.elegans, Arabidopsis and Aspegillus genomes.
The result of the analysis is presented in EMBL-like format and by using a special graphical interface to display the gene structure predicted in Internet.
2.11. Results representation
The programs results can be visualize by using the java applet “FeatureView” implemented in JAVA and can be used for the interactive evaluation of the gene model predicted (Fig. 1). The WebGene system is available on the Internet at the ITBA-CNR Web Server (http://www.itba.mi.cnr.it/webgene).
Acknowledgements
This work was supported by the EC project BIO4-CT95-0226 and the Italian CNR and Russian “Human Genome Project”.
References
- Milanesi and I.B. Rogozin “Prediction of human gene structure. In: Guide to Human Genome Computing (2nd ed.) (Ed. M.J.Bishop), Academic Press, Cambridge, 215-259. (1998).
- Milanesi, M. Muselli, P. Arrigo “Hamming Clustering method for signals prediction in 5′ and 3′ regions of, eukaryotic genes” Comput. Applic. Biosci, 12 (5) p399-404 (1996)
- Rogozin and L. Milanesi “Analysis of Donor Splice Sites in Different Eukaryotic Organisms” J. Mol. Evol. 45, 50-59 (1997).
- Rogozin, L. Milanesi, N.A. Kolchanov “Gene structure prediction using information on homologous protein sequence” Comput. Applic. Biosci., 12, 161-170 (1996)
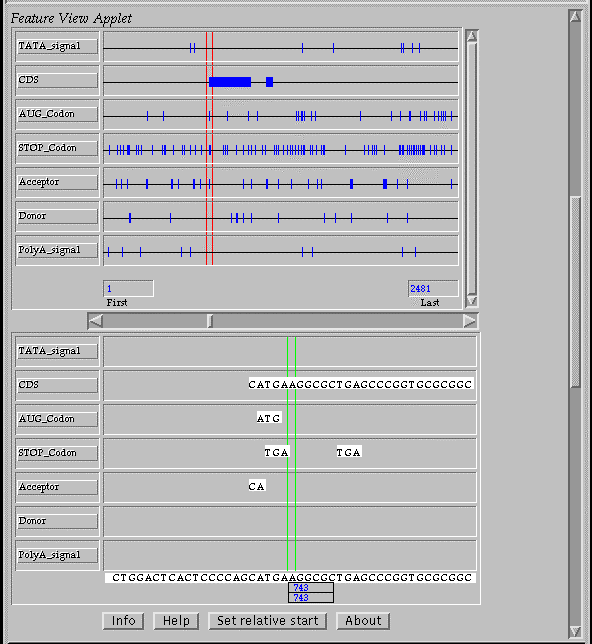
Figure 1. The queries and the programs results are visualized on Netscape and Explorer browsewrs by using the java applet “FeatureView”.