KOLPAKOV F.A.+, FROLOV A.S., PONOMARENKO M.P., PODKOLODNY N.L.1
Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, 10 Lavrentiev Ave., Novosibirsk, 630090, Russia;
1Institute of Computational Mathematics & Mathematical Geophysics, Siberian Branch of the Russian Academy of Sciences, 6 Lavrentiev Ave., Novosibirsk, 630090, Russia;
+Corresponding author
Keywords: Internet, query retrieval language, databases, viewer, protein, structure, function, anthology
Any biological functions are realized as a result of interactions between the 3D structures DNA, RNA, proteins, and low-weigh molecules. The protein regions contacting these molecules are called “active centers” in the 3D-structure level and “functional sites” in the sequence level. That is why each of proteins has a notably large number of attributes necessary to be determined for their more or less objective description. For example, a fixed protein can be characterized by its biological function, its signal peptide, active centers, disulfide bonds, 3D-, secondary, supersecodary and domain structures, amino acid sequence, the gene coding for this sequence, location of this gene on DNA, regulatory regions of this gene, the functional sites constituting these regulatory regions, the protein factors switching on this gene using these signals, and in turn, these protein factors can have their functions, structures, genes, regulations, etc. The totality of these and hundreds of other (evolutionary, physiological, etc.) attributes of a protein constitutes its onthology which is the best starting point for any protein query retrieval language comprehensible for a user searching any data on this protein through all the Web-available databases.
The protein 3D structures X-ray identified, and also, their signal peptides, active centers, 3D-, secondary, supersecodary and domain structures are compiled in the Brookhaven Protein Data Bank; their sequences decoded from genes in the EMBL nucleotide sequence database (Stoesser, 1998), GenBank Database (Benson, 1998), Data Bank of Japan on Genome Sequence Data (Tateno, 1998), Genome Sequence DataBase GSDB (Harger, 1998), Saccharomyces Genome Database SGD (Cherry, 1998), Drosophila database Flybase (Flybase Consortium, 1998), Human Genome Database GDB (Letovsky, 1998), the database Virgil (Achard, 1998), and those sequenced directly in SWISS-PROT and PIR databases; evolutionary variants of these protein sequences, in the databanks HSSP, PROFILE, and PROSITE; and, then, location of the protein binding sites on DNA molecules in the Eukaryotic Promoter Database EPD (Cavin, 1998), the database EpoDB of genes expressed during vertebrate erythropoiesis (Salas, 1998), and the databases on transcription regulation TRANSFAC (Wingender, 1997), TRRD (Kel A, 1997), and COMPEL (Kel O, 1997); values of the protein/DNA affinity in the database ACTIVITY (Ponomarenko, 1997); and significant patterns of the structural organization of these regions and sites, in the Object-Oriented Transcription Factors Database OOTFD (Ghosh, 1998), ConsInspector (Frech, 1997), MatInd and MatInspector (Quandt, 1995), GenomeInspector (Quandt, 1996), ModelInspector (Frech, 1996), TRANSFAC (Wingender, 1997), CoreSearch (Wolfertstetter, 1996), TESS (Schug and Overton, 1997), MATRIX SEARCH (Chen, 1995), and, finally, the network schemes in which the proteins interacting one another and, also, DNA, RNA and low-weigh molecules to form the molecular singal transduction passways are compiled within the database GeneNet (Kolpakov, 1998), etc. Over 400 molecular genetic databases are currently Web-available; however, they are virtually completely separate and have their own formats, query languages, input/output systems, etc. This disconnection of experimental data on proteins corresponds to the information technology level of 20 years ago, when the accumulation of these data just started. The lack of connection of the experimental data on the protein 3D structures and functions is the main obstacle to plan objectively the research into these macromolecules. Nevertheless, the proteins function only within complex networks composed by many hundreds of intermolecular relationships coordinated spatial-temporally between one another within the entire living organism. These so-called genetic networks are so multi-component, cumbersome, intricate, and complex that no experimental methods for their investigation as a whole are yet available.
Recently appeared INTERNET allows the connectivity of all these experimental data on proteins to be reestablished via integrating the databases containing all the data. Thus, it becomes, indeed, one of the crucial problems of the protein bioinformatics. For example, the Brookhaven Protein Data Bank (Bernstein, 1977) contains the 3D structures of human immune proteins, interferons and immunoglobulins and the core proteins of pathogenic viruses. These 3D structures are completely unconnected. In turn, the EMBL nucleotide sequence database (Stoesser, 1998) and GenBank database (Benson, 1998) contain the information on the genes of the human immune and viral pathogenic proteins, which are unconnected too. In addition, the databases EPD (Cavin, 1998), EpoDB (Salas, 1998), TRANSFAC (Wingender, 1997), TRRD (Kel A, 1997), and COMPEL (Kel O, 1997) contain the information on switching on these human and viral genes, that are unconnected too. However, the GeneNet database (Kolpakov, 1998) describes only the above protein-proteint and protein-DNA interactions specially-temporary coordinated within the gene network of human interferon-induced immune response to the viral infection.
This GeneNet has beens developed especially for computable description of the semantic relationships between the biological entities constituting the living organism in the process of their functioning contains the whole metainformational part of protein onthology. That was, then, complemented by the protein genetic query retrieval language MGL-Prot (Kolpakov, 1997) designed for searching protein biological and genetic databases for the information of interest. This MGL-Prot language has two molecular-genetic object-oriented parts: (1) interactive graphic user interface for visualization of macromolecules and (2) the class library and the high-level coding language based on usage of the genetic terms and their synonyms. These both were exiended by (3) the object-oriented description specified for the Web-available databases having the protein attributes. That are resulting the antology-based integrator for the protein databases. Figure is showing schematically how it can be performed via the three types of the MGL-based interconnections, respectively:
- User<=>MGL-Viewer,
- MGL-Viewer<=>MGL-ClassLibruaries,
- MGL-ClassLibruaries<=>MGL-Prot.
In this three step way, the MGL-Prot query retrieval language for the protein onthology-based searching through the PDB, EMBL, TRRD, TRANSFAC, SWISS-PROT, MEDLINE databases on various proteins.
We are grateful to Ms. Galina Chirikova for the help in translation. This work was supported by Russian National Human Genome Project, Russian Ministry of Science and Technical Politics, Siberian Branch of the Russian Academy of Sciences (grant No. IGSBRAS-97N13), and Russian Foundation for Basic Research (grants Nos. 96-04-50006, 97-07-90309, 97-04-49740, 98-07-90126).
Reference
- Kolpakov, F.A., Ananko, E.A., Kolesov, G.B. and Kolchanov N.A. (1998), “GeneNet: a database for gene networks and its automated visualization” Bioinformatics, 14, accepted in press.
- Kolpakov, F.A., Babenko, V.N. (1997) Computer system MGL: a tool for sample generation, graphical representation, and analysis of genomic regulatory sequences, Mol. Biol., 31, 647-655.
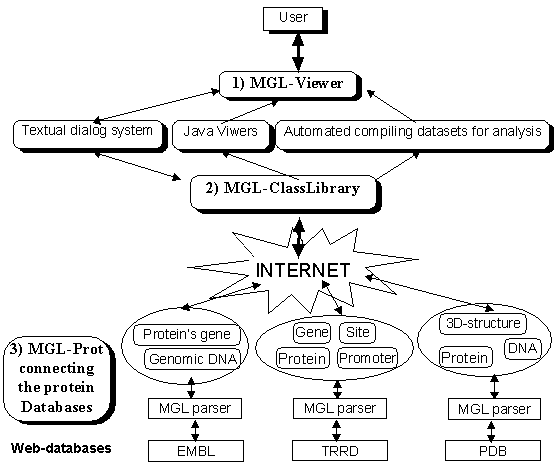 |
Figure. Scheme of the MGL-Prot query retrieval language for the protein onthology-based searching for their 3D-structures and functions in the Web-available databases. |