VTYURIN N.+, BATURIN V., GULIN V., GORYACHEV N.
Institute of Molecular Genetics, Russian Academy of Sciences, Kurchatov square, Moscow, 123182, Russia;
e-mail: N.Vtyurin@g23.relcom.ru
+Corresponding author
Keywords: protein, pair structural alignment, dynamic programming
Abstract
We present the computer program 3D_ALIGN for 3D pare alignment of two protein 3D structures. The program does not need any sequence gomology and any previous assumption about 3D structure similarities of starting proteins. The program can determine itself the degree of 3D similarity. The output of this program is 3D coordinates of second proteins rotated and shifted to the best fitting to coordinates of the first protein. You can see and manipulate stereoscopic image of starting structures and final result (both proteins). The second kind output of this program is the linear representation of 3D alignment and the list of structure conservative regions that fit to given RMS deviation. It is possible to use this program for the purpose of computer modelling and 3D classification of protein 3D structures. The program was developed for IBM compatible computers, needs only 466 Kb hard disk space and can work even from floppy drive.
Introduction
The huge gap between number of known protein primary structures (200’000) and known 3D structures of protein molecules (6’000), which increases each year, makes especially actual development effective computer modeling methods of 3D structure of proteins based on their amino acid sequence. One of the most important tools to solve this problem are 3D alignment of tertiary protein structures programs. Such programs are necessary for classification of known 3D structures. To search for investigated protein with known amino acid sequence analogue or analogues in bank of already known 3D structures, it is necessary at first to separate all contents of 3D bank to corresponding “shelves” (families of 3D structures). After the analogues are found, the program of 3D alignment is necessary to search structure conservative regions (SCR) of these analogues and evaluate of root-mean-square (RMS) deviation on these SCR between analogs.
There are known some good programs of such kind [1-3]. For example it is the automatic 3D alignment part of program SUPPOS in program package WHAT_IF (Gert Vriend, Heidelberg, Germany) and program COMPOSER from the package SEBYL of firm TRIPOS (Authors: group of T.Blundell, London, England). When we have decided to write our own program of 3D alignment, main purpose, which we set ourselves a target, was more complete understanding of algorithms of such kind of programs, and possibility to change the program at any moment, for check our own ideas and hypotheses of other authors and possible improvement of such program. The second purpose was creation compact (pocket) program, which could work on more widespread IBM compatible computers (Notebooks), and which can be used even in the house.
Methods and algorithms
The input files of program 3D_ALIGN are two any Brookhaven files. At first program performs the analysis of subunit contents in both files and enables to choose for alignment any two subunits from all subunits of both files. Then program performs scanning 3D structure of the first subunit relatively to another one to search the best starting superimposed position for the beginning of 3D alignment itself. At first. 3 N terminal amino acids of first subunit matches with 3 C terminal amino acids of second subunit and program performs superposition of matched amino acids C-alpha atoms, calculates the D=(RMS)/(Lp) value for this superposition, where “RMS” is Root Mean Square of matched amino acids, “L” is the number of matched amino acids, “p” is the power of L. The default value of “p” is 1. Then 4 N terminal amino acids of first subunit matches with 4 C terminal amino acids of second subunit and program superimposes for this match and calculates value of D for this superposition. This procedure repeats up to match where 3 C terminal amino acids of first subunit matches with 3 N terminal amino acids of second subunit. After this program finds from this set of matches the match with minimum value of D. The superposition of this match becomes starting superposition for the process of alignment itself. User can change the value of “p” to change the sense of 3D alignment, namely more rough but global 3D alignment or more precise but local 3D alignment.
After this for starting superimposed position of subunit 3D structures program generates matrix of distances between corresponding C-alpha atoms of both subunits. Then, using method of dynamic programming, similar algorithm of Needlman Wunsh [4], program finds the optimal way through this matrix with the minimum score of distances between amino acids. On the base of the results of this procedure program generates new set of matched amino acids excludes from this set matches with distances between C-alpha atoms of corresponding amino acids more than 3.0 angstrems. Then program superimposes two structures on the base of new set of matches and generates new matrix of distances and so on. The procedure of finding correspondence of amino acids and their superimposition are iteratively repeats so long as after the next iteration the list of corresponding amino acids not changed. This procedure allows to find structure conservative regions of two subunits.
The output of this program is Brookhaven format file with 3D coordinates of second proteins rotated and shifted to the best fitting to coordinates of the first protein. One can see and manipulate stereoscopic image (with mirror stereoglasses) of starting structures and final result (both proteins). The second kind output of this program is the linear representation of 3D alignment and the list of structure conservative regions that fit to given RMS deviation.
The program was written by language C ++.
Process of two proteins alignment: (Cytochrome C550 (155C), length 135 amino acids residue and Cytochrome C5 (1CC5), length 83 amino acids residue) takes on IBM compatible computer (486DX-2, 80 MHz) about 1 minute [5].
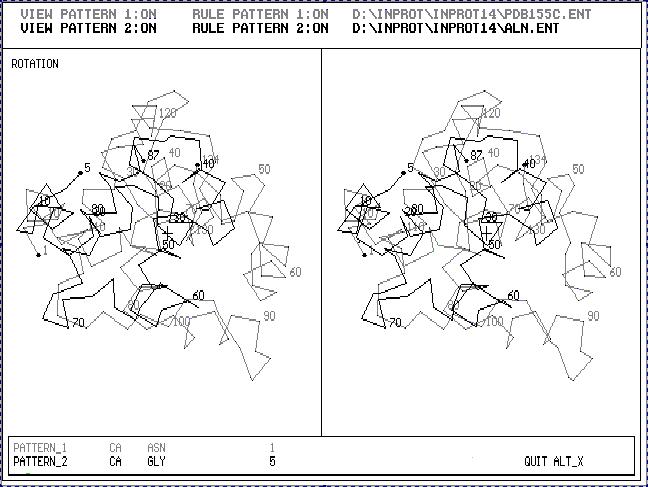
Fig.1. Stereoscopic image of the result of 3D_ALIGN program applied to Cytochrom C550 (black one) and Cytochrom C5 (gray one). These are proteins with enough different 3D structures. The sequence gomology of these two proteins is about 20%.
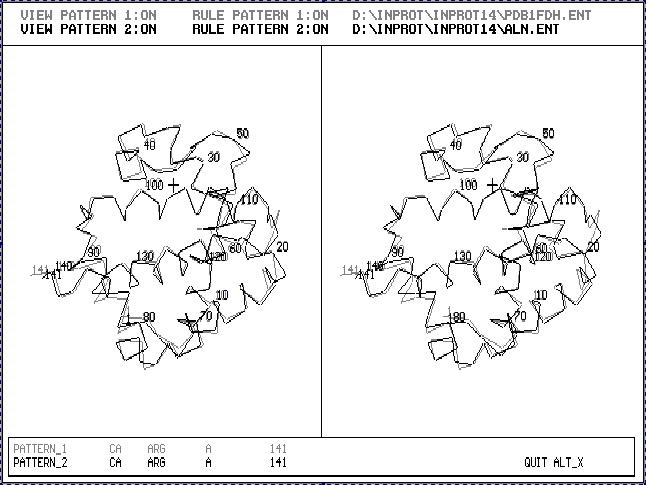
Fig.2. Stereoscopic image of the result of 3D_ALIGN program applied to Human Fetus Hemoglobin (black one) and Human Hemoglobin (gray one). This is an example of proteins with very similar 3D structures.
Acknowledgements
This work was supported by Russian Fundation for Basic Research. Grants ı 94-07-20442 and
ı 96-04-49407.
References
- G. Vriend, “WHAT IF”: A Molecular Modelling and Drug Design Program” Journal of Molecular Graphics 8, 52-56 (1990)
- A. Sali, J.P. Overington, M.S. Johnson, T.L. Blundell, “From Comparisons of Protein Sequences and Structures to Protein Modelling and Design” TIBS 15, 235-240 (1990)
- “SYBYL Molecular Modelling Software” TRIPOS Associates, Inc St. Louis, Missouri, USA. (1994)
- S.B. Needleman, C.D. Wunsch, “A General Method Applicable to the Search for Similarities in the Amino Acid Sequence of Two Proteins” J. Mol. Biol. 48, 443-453 (1970)
- N. Vtyurin, V. Baturin, V. Gulin, A. Shplatov, V. Katalov, A. Katalov, “The First Version of Integrated Protein Database IN_PROT for IBM PC Compatible Computers with Some Computer Modelling Tools” Protein Science 6, suppl.1, 75 (1997)