KOLESOV G.B., KOLPAKOV F.A.+, KOLCHANOV N.A.
Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, 10 Lavrentiev Ave., Novosibirsk, 630090, Russia;
e-mail:fedor@bionet.nsc.ru
+Corresponding author
Keywords: transcription regulatory regions, modular structure, pairwise and multiple alignment, similarity profiles
Abstract
It is known that transcription regulatory regions have a modular structure and contain transcription factor binding sites, composite elements, enhancers, silencers, and other functional modules. The situation when the same functional module is located in different positions relative to the transcription start. We propose to solve this problem by combining the methods of pairwise and multiple alignment with analysis of the similarity profiles of the sequences.
The promoters of interferon-induced and erythroid-specific genes were investigated. Analysis of similarity profiles for this group promoters reveals that their have large number of similar motifs. The motifs revealed on promoters of interferon-induced genes were subjected to multiple alignment using Gibbs algorithm and was shown their correspondence to binding sites of transcription factor regulating antiviral response.
1. Introduction
It is known that transcription regulatory regions have a modular structure and contain transcription factor binding sites, composite elements, enhancers, silencers, and other functional modules. The specificity of the function of a transcription regulatory region is determined by both the presence of certain modules and their location relative to the transcription start of the gene and to one another. The situation when the same functional module is located in different positions relative to the transcription start or encompassed by different other modules is typical. This hinders essentially the revealing of the functional modules constituting promoters and other long regulatory regions, as the conventional methods of multiple alignment are inapplicable in this situation. We propose to solve this problem by combining the methods of pairwise and multiple alignment with analysis of the similarity profiles of the sequences.
2. Algorithm
Letís consider a set of N sequences of certain regulatory region and find for the sequence Si (i=1..N) the pairwise local alignments (Waterman, Eggert, 1987) with the sequence Sj (j=1..N, j<>i). Only the alignments that has the score exceeding the threshold level are chosen. If several alignments are overlapping, the best of them is chosen.
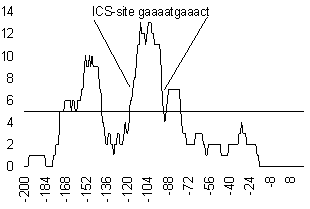
Figure 1. Similarity profile of the promoter of interferon-inducible gene 6-16.
The local alignments obtained through this procedure are used to construct an integral similarity profile of the i-th sequence with all the others:
![]()
where ![]() (k) is the number of the local alignments between the i-th and j-th sequences containing the k-th position of the i-th sequence. The value F(i,k) calculated in this way corresponds actually the number of similar fragments in the other sequences (independently of their location) to the k-th position of the i-th sequence. The maximums of F(i,k) correspond to those regions of the i-th sequence that have many similar regions in the other sequences of the set in question, that is, to potential functional motifs.
(k) is the number of the local alignments between the i-th and j-th sequences containing the k-th position of the i-th sequence. The value F(i,k) calculated in this way corresponds actually the number of similar fragments in the other sequences (independently of their location) to the k-th position of the i-th sequence. The maximums of F(i,k) correspond to those regions of the i-th sequence that have many similar regions in the other sequences of the set in question, that is, to potential functional motifs.
The following approach was used to estimate the statistical significance of the maximums of the profile obtained. Multiple generation of the sets of random sequences Sj (j=1..N, j<>i) with the same nucleotide frequencies as in the real sequences was performed. Each random sequence from the set Sj was aligned with the sequence Si, and the profile of total similarity Frand(i,k) was constructed. The distribution of the values Frand(i,k) was considered Poisson, and the k* was found at a fixed i, so that the probability P(Frand (i,k) > k*) < 0.05.
Only those regions of the profile where the condition F(i,k)>k* was fulfilled were considered significant (Fig. 1). The corresponding regions of the sequence Si and the similar regions of the sequences Sj were considered the regions that correspond to the potential functional modules abundant in the sequence set under study. An example of location of the motifs revealed by this method is represented in Fig. 2.
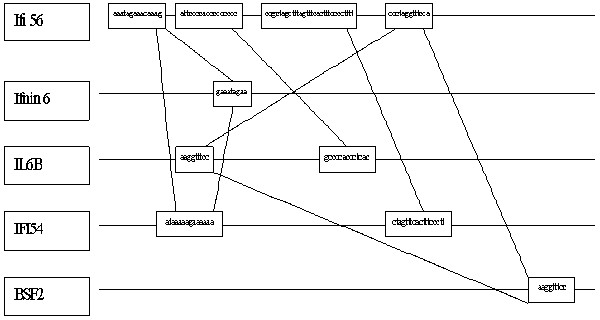
Figure 2. Examples of similarity motif disposition within promoters of the interferon inducible genes.
Then the group of motifs revealed by this procedure is subjected to multiple alignment using Gibbs algorithm (Lawrence et al., 1993). This allows to reveal a finer structure of their similarity (Fig. 4). It is possible to relate the significant motifs found to known functional sites. In particular, the region of the highest maximum in Fig. 1 corresponds to the known ISG factor binding site, specific for interferon-regulated genes.
Note that in the groups of regulatory genomic sequences with a similar function, the motifs that have a similar location in a number of sequences may exist. To reveal such regions, the integral similarity profile for the entire set of RGS is constructed as follows: F(k) = ![]() .
.
3. Results
The integral similarity profiles constructed for three sets of promoters (every set is free from homologous sequences):
- 21 promoters ( region [-200, +20] relative the transcription start site) of human interferon-induced genes compiled on the base information from the GeneNet database (Kolpakov et.al., 1998; http://wwwmgs.bionet.nsc.ru/systems/Mgl/GeneNet/);
- 21 promoters ( region [-200, +20]) of different human genes (expressed in different cells and under different conditions) compiled from EPD database;
- 10 promoters (region [-200, +60]) of erythroid-specific genes from different spcies (http://wwwmgs.bionet.nsc.ru/Dbases/NSamples/auto1.exe).
The regions of similarity are revealed by comparison of the profile F(K) with the corresponding profile calculated for the set of random sequences with the same nucleotide content (Fig. 3).
Characteristic of the interferon-regulated gene promoters is a regular exceeding of the integral similarity profile compared to the corresponding random profile; the region [-180;-60] exhibited the maximal surpassing over the random levels.
Four regions of surpassing over the random profile were recorded in the integral similarity profile of erythroid-specific promoters. One corresponds to the region of recruitment of the basal transcription complex [-30;-10]; the second [-90,-50] coincides with the characteristic location of CCAATT box. The rest two, [-110,-130] and [-170, -190], correspond to some other functional motifs frequently occurring in these regions of erythroid-specific promoters.
As for the promoters of different human genes their integral similarity profile not exceeds significantly the corresponding profile for random nucleotide sequences. This is support the significance of revealed similarity for promoters of interferon-regulated and erythroid-specific genes.


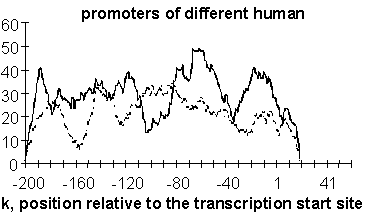
Figure 3. Integral similarity profiles of gene-specific groups of promoters.
The motifs revealed on promoters of interferon-induced genes were subjected to multiple alignment using Gibbs algorithm (Lawrence et al., 1993). This enabled to reveal a finer structure of their similarity (Fig. 4). Thus revealed motifs correspond to binding sites of transcription factor regulating antiviral response (Anan’ko et.al., 1997).


Figure 4. Multiply alignment of similarity motifs by Gibbs sampling. Core of the alignment coincides with the known binding site of ISG factor (specific regulator of interferon-inducible genes).
Acknowledgements
We are grateful to Yury Kondrakhin for valuable discussions and to Ms Galina Chirikova for assistance in translation.
This work was supported by grants from the Russian Foundation for Basic Research (No.97-04-49740, 97-07-90309, 96-04-50006, 98-04-49479, 98-07-90126); Russian Ministry of Science and Technologies; Russian Human Genome Project; Russian Ministry of High Education.
References
- E.A. Ananíko, S.I. Bazhan, O.E. Belova and A.E. Kel “Mechanisms of transcription of the interferon-induced genes: a description in the IIG-TRRD information system” Mol. Biol. (Mosk)., 31, 592-605, 1997.
- M.S. Waterman, M. Eggert “A new algorithm for best subsequence alignments with application to tRNA-rRNA comparisons” J. Mol. Biol., 197(4), 723-728, 1987.
- C.E. Lawrence, S.F. Altschul, M.S. Boguski, J.S. Liu, A.F. Neuwald, J.C. Wootton “Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment” Science, 262, 208-214, 1993.
- F.A. Kolpakov, E.A. Ananko, G.B. Kolesov, and N.A. Kolchanov, “GeneNet: a database for gene networks and its automated visualization through the Internet” Bioinformatics, 14(6), in press, 1998.