Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, 10 Lavrentiev Ave., Novosibirsk, 630090, Russia;
e-mail:fedor@bionet.nsc.ru
+Corresponding author
Keywords: molecular genetics databases, nucleotide sequence samples, nucleotide sequences analysis, visualization
Abstract
The object-oriented computer system MGL is designed for (1) searching for and extracting the information from molecular genetics databases; (2) automated generation of nucleotide sequence samples of various gene components basing on semantic analysis of the EMBL FEATURE TABLE information and samples of promoters and transcription factor binding sites basing on the information from the EPD and TRRD databases, respectively; (3) nucleotide sequences analysis; and (4) visualization of the database-contained information and the results of analyses performed.
The core of the MGL is the class library based on the idea that the set of classes corresponds directly to the main concepts of molecular genetic data. A specialized high-level object-oriented language allows a user to generate samples and analyze nucleotide sequences in an automatic mode. The MGL system has a friendly user interface designed for Windows. The MGL demo version is available at http://wwwmgs.bionet.nsc.ru/systems/Mgl/mgl.html.
1. Introduction
Research into the mechanisms, organization patterns, and functions of the genome is one of the main goals of molecular biology. Large-scale projects of genome sequencing are being performed to achieve this goal. The data obtained on the primary nucleotide sequences are being accumulated in the EMBL and GenBank databases. In addition, specialized databases are being developed, such as EPD, TRRD (Kel, A.E. et al., 1997), TRANSFAC (Wingender et al., 1996), and COMPEL (Kel O.V. al., 1995), providing a detailed description of various peculiarities of the regulation of gene expression. Currently, over 400 specialized databases on molecular biology and genetics, accessible via the Internet (http://www.infobiogen.fr/services/dbcat/), exist in the world. Hence, an efficient search for the information of interest is becoming more and more crucial.
One of the major problems in computer analysis of genomic sequences is the absence of representative samples of various nucleotide sequences along with the tools for their automated generation. By a sample is meant a group of nucleotide sequences or their fragments possessing certain biological function. The EMBL database is used as a basic source of information for generating the representative samples of nucleotide sequences of various gene regions (exons, introns, protein-coding regions, 5′ and 3′ untranslated regions, etc.) and functional sites (promoters, splicing sites, polyadenylation sites, etc.). The EMBL database contains all the currently decoded nucleotide sequences along with the information on their structure-function organization. However, this information is poorly formalized, can be incomplete and erroneous. Hence, the semantic analysis and accurate examination of the data on the structure-function organization of nucleotide sequences are required previously to generating the sequence samples in an automated mode.
Numerous methods for analysis of nucleotide sequences have been so far developed in the world; as a rule, they are realized as separate programs or software packages. However, the majority of these methods does not support the operation with databases and lack the developed tools for graphical representation of the results obtained. These shortcomings limit considerably their efficiency and application area.
Comparison of the results obtained by analysis of nucleotide sequences can increase drastically the accuracy of the analysis performed, accuracy of the predictions of the structure and function of genes, their expression patterns, etc. It is of the utmost importance now, when a tremendous number of genomic fragments with unknown structure and function is being sequenced.
Thus, to solve these problems, an integrated computer system provided with powerful graphic user interface is required to provide the user with the tools for simultaneous operation with a number of databases via the Internet, automated sample generation, their comprehensive analysis, and graphical representation of the data from databases and the results obtained.
2. The structure of the MGL system
The computer system MGL (Kolpakov, Babenko, 1997) consists of the three sections: (1) an object-oriented class library; (2) a specialized high-level object-oriented language; and (3) a user interface.
The object-oriented class library. The main classes of this library can be divided into five groups: (1) the classes corresponding to the basic molecular genetic notions (Sequence, Site, GeneStructure, etc.); (2) the classes for operation with databases; (3) for generating samples of genomic sequences; (4) for analyzing genomic sequences; and (5) for graphical representation of the data from databases and the results obtained. The library is realized in C++; in addition, sections 1, 2, and 5 are also realized in Java and used to develop the applications for Internet-based visualization of gene networks within the GeneNet database (Kolpakov et. al, 1998) and transcription regulatory regions within the TRRD database (Kel A.E. et al., 1997).
Molecular Genetics Language (MGL) is a specialized high-level object-oriented language. It has special types corresponding to the basic molecular genetic notions (for example, SITE, SEQUENCE, SITE_SET, SEQUENCE_SET, etc.), the notions connected with database operation (DATABASE, ENTRY, ENTRY_SET, etc.), and corresponding to certain types of analysis (ALIGNMENT, PROFILE). An example of the MGL program is shown in Section 4.
Graphic user interface is realized for Windows. It contains the editor for MGL programs, the windows for viewing results in textual and graphical forms, and a set of dialogues for generating samples of nucleotide sequences and their analysis.
3. Possibilities and characteristics of the MGL system
3.1. Database access
Computer system MGL provides the access (search for and extraction of information) to the databases (1) accessible via the SRS (Sequence Retrieval System; Etzold, Argos, 1993) and (2) installed in the user’s computer (EMBL, ENZYME, EPD, PROSITE, SWISS-PROT, etc.). In addition, the MGL systems supports the operations with various samples generated from databases.
3.2. Automated generation of nucleotide sequence samples
MGL provides for the automated generation of the nucleotide sequence samples. The samples of various genes components (promoters, introns, exons, splicing sites, polyadenylation sites, etc.) are generated basing on semantic analysis of the FEATURE TABLE of the EMBL database. The semantic analysis includes checking the information for compliance with the basic principles of the eukaryotic gene organization. The system MGL provides the user with two modes of sample generation: (1) completely automatic and (2) allowing the errors to be manually corrected. In the first mode, the system will try to correct the error in the gene structure description by itself basing on the available knowledge; in the second case, the user corrects the gene structure description, and then the system analyzes it again.
While generating the samples of promoter regions and transcription factor binding sites, the system MGL uses the EMBL database as a source of nucleotide sequences and the EPD and TRRD databases as a source of the data on location of the corresponding functional sites in these sequences.
The nucleotide sequence samples generated can be stored in different formats (EMBL-like, FASTA, PIR, etc.). In addition, the system MGL provides an easy conversion from one format to another.
3.3. Methods for nucleotide sequences analysis
The computer system MGL contains a wide range of tools for analysis of nucleotide sequences including calculation of nucleotide and oligonucleotide compositions; pairwise general and local alignments; rapid estimation of pairwise general alignment significance (Seledtsov and Kolpakov, 1998); calculation of the number of synonymous and nonsynonymous substitutions; analysis of leader sequences (Kochetov et. al., 1998); calculation of similarity profiles for groups of functionally related sequences basing on their pairwise local alignments (Kolesov and Kolpakov, 1998); search for transcription factor binding sites (Kel A.E. et. al, 1995), etc.
3.4. Visualization of the information from databases and the results obtained
The system MGL allows also the information from databases and the results obtained to be represented graphically. For example, the data from EMBL are represented as a scheme of the structure of the gene and its functional sites; the data from TRANSFAC and TRRD, as graphical maps of gene regulatory regions. Various functional sites found as a result of the analysis performed can be also represented as a graphical map.
3.5. Automated recording of operation
Automated recording of all the steps of operation is an important characteristic of the MGL system. For this aim, the system creates a special file, serving as a working logbook. This file contains the dates of generation of the samples and performance of the analyses of nucleotide sequences, names of the functions and their options used, names of the files containing the samples generated and the results obtained, messages of the system in the course of operation, etc. This gives the user a complete information on the process of automated sample generation and sequence analysis.
4. An example of the MGL program
An example of the MGL program generating the sample of human promoters basing on the data from EPD and EMBL and subsequent searching for potential transcription factor binding sites in these sequences is shown below, supplemented with comments. The results obtained can be represented in both the textual and graphical forms (Fig. 1)
// Connecting the system MGL with the databases EPD and EMBL, installed in the user’s computer
DATABASE dbEPD = DB_Connect ( “EPD” );
DATABASE dbEMBL = DB_Connect ( “EMBL” );
// Searching the EPD databases for the entries containing the information on human promoters
ENTRY_SET entries = DB_Search ( dbEPD, “OS=human” );
//Generating the sample of human promoters [-200;+20] relative to the transcription start
SEQUENCE_SET sequences = CreatePromotersEPD ( entries, dbEMBL, 200, 20 );
// Saving of the sample into a file in a FASTA format
Save( sequences, F_FASTA, “prom.set”);
//Searching for the sites according to the consensus: “cons.dbf”, name of the file with consensuses; “C”, consensus method is to be used for the search; 2, the search is to be performed in both strands; 10, per cent of mismatch with the consensus
SITES_SET sites = SitesFind( sequences, “cons.dbf”, “C”, 2, 10);
// Saving the results obtained in a file in a text form
SitesSave( sites, “sites_Ò.txt” );
// Graphical representation of the sites found for s specified gene
ViewSitesGene(sites, “Hs hsp 70K”);
// Saving of the resulting image in a file in the Windows Metafile format
SavePicture (“sites.wmf”);
Acknowledgements
We are grateful to Yury Kondrakhin for valuable discussions and to Ms Galina Chirikova for assistance in translation.
This work was supported by grants from the Russian Foundation for Basic Research (No.97-04-49740, 97-07-90309, 96-04-50006, 98-04-49479, 98-07-90126); Russian Ministry of Science and Technologies; Russian Human Genome Project; Russian Ministry of High Education.
References
- T. Etzold and P. Argos, “SRS – an indexing and retrieval tool for flat file data libraries” CABIOS, 9, 49-57, 1993.
- A.E. Kel, Y.V. Kondrakhin, F.A. Kolpakov, O.V. Kel, A.G. Romaschenko, E. Wingender, and
N.A. Kolchanov, “Computer tool FUNSITE for analysis of eukaryotic regulatory genomic sequences” (Proc. III Intern. Conference of Intelligent Systems for Molecular Biology, California, US), 197-205, 1995. - A.E. Kel, N.A. Kolchanov., O.V. Kel,` A.G. Romashchenko, E.A. Ananíko, E.V. Ignatíeva, T.I. Merkulova, O.A. Podkolodnaya, I.L. Stepanenko., A.V. Kochetov, F.A. Kolpakov, N.L. Podkolodny, and A.N. Naumochkin, “TRRD: database on transcription regulatory regions of eukaryotic genes” Mol. Biol. (Mosk.), 31, 521-530, 1997.
- O.V. Kel, A.G. Romaschenko, A.E. Kel, E. Wingender, and N.A. Kolchanov. “A compilation of composite regulatory elements affecting gene transcription in vertebrates” Nucl. Acids Res., 23, 4097-4103, 1995.
- A.V. Kochetov, M.V. Pilugin, F.A. Kolpakov, V.N. Babenko, E.V. Kvasnina, and V. K. Shumny, “Structural and compositional features of 5í untranslated regions of higher plant mRNAs”, (Proc. I Intern. Conference on Bioinformatics of Genome Regulation and Structure, Novosibirsk, Russia), 1998.
- G.B. Kolesov and F.A. Kolpakov. “New method for the study of the modular structure of transcription regulatory regions” (Proc. I Intern. Conference on Bioinformatics of Genome Regulation and Structure, Novosibirsk, Russia), 1998.
- F.A. Kolpakov and V.N. Babenko. “Computer system MGL: tool for sample generation, visualization and analysis of regulatory genomic sequences” Mol. Biol., 31, 540-547, 1997.
- F.A. Kolpakov, E.A. Ananko, G.B. Kolesov, and N.A. Kolchanov, “GeneNet: a database for gene networks and its automated visualization through the Internet” Bioinformatics, 14(6), in press, 1998.
- I.A. Seledtsov and F.A. Kolpakov. “Rapid estimates of statistical significance of the pairwise nucleotide sequence alignment” (Proc. I Intern. Conference on Bioinformatics of Genome Regulation and Structure, Novosibirsk, Russia), 1998.
- E. Wingender, P. Dietze, H. Karas, and R. Kneuppel. “TRANSFAC: a database on transcription factors and their DNA binding sites” Nucl. Acids Res., 24, 238-241, 1996.
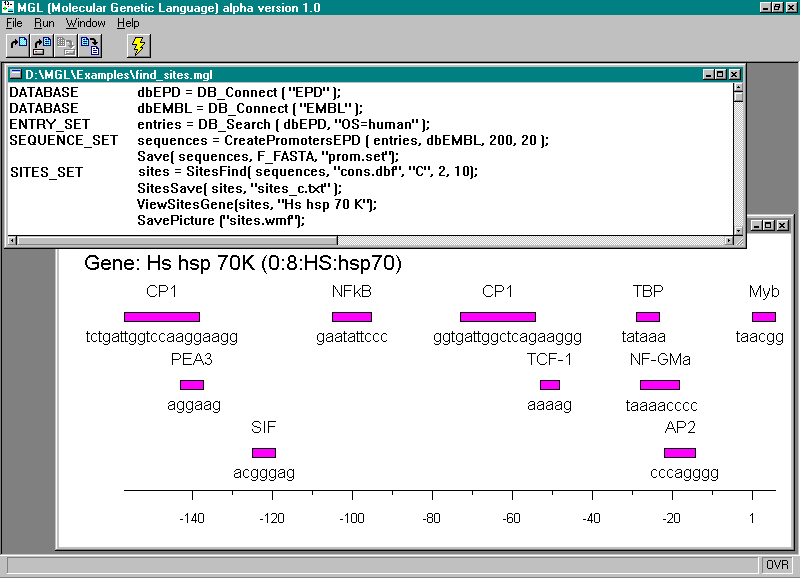
Figure 1. Operation of the system MGL. The upper window contains the text of the program; the lower, the map of the transcription factor binding sites found for a specified gene.