
Instructions to Authors
Our rules for preparation of extended abstracts are similar to rules recommended for papers in BIOINFORMATICS with some modifications.
Extended Abstracts must be written in English and Abstract length should not exceed 4 pages.
Please use 12-point Times New Roman font. Do not indent the start of paragraphs. Text should be single-spaced with 3.5 cm from the left margin and 2,5 from other margins.
Please subdivide manuscripts of extended abstract into the following sequence of sections:
Title.
The Title should be typed in bold capital letters (14-point Times New Roman font). Separate title from names of authors by one blank line.
Names of authors.
The surname and initials of each author should be followed by his/her department, Institution, city, postal code, country and E-mail address. Any changes of address may be added to the footnotes. The corresponding author be indicated by an asterisk and the footnote ‘To whom correspondence should be addressed’.
Separate names of authors from key words by one blank line.
Key words.
Up to ten key words should be supplied to assist with the compilation of the Subject Index.
Structured Resume.
Resume is structured with a standard layout such that the text is divided into sub-sections under the following three headings: Motivation, Results, Availability. In cases where authors feel the headings inappropriate, some flexibility is allowed. The abstracts should be succinct and contain only material relevant to the headings. A maximum of 150 words is recommended. If internet hyperlinks are available for any part of the abstract, then this should be given in the form of ‘clickable text’, i.e.{{http://www…}}.
Motivation: This section should specifically state the scientific question within the context of the field of study.
Results: This section should summarize the scientific advance or novel results of the study, and its impact on computational biology.
Availability: This section should state software availability (academic or commercial) if the paper focuses mainly on software development or on the implementation of an algorithm. Examples are: free availability over the internet on WWW or ftp server; available on request from the authors; or available as a commercial package. The complete address (URL) should be given.
Introduction
Methods and algorithms
Implementation and results
Discussion.
References.
Abstracts should be submitted in an electronic form as attachment files in MS Word v.6-7. Figures (black and white, not colour) should be submitted individually as JPEG, GIF, BMP or PNG bitmaps. They must be named as Fig.1, Fig.2 and so forth. Legends of figures should be placed within the text at locations where they will appear in the abstract.
Receipt of abstracts will be acknowledged by the Organizing Commitee.
Nikolay A Kolchanov,Please, submit manuscript including figures as an E-mail attachment to
Institute of Cytology and Genetics
Novosibirsk, Russia
E-mail: kol@bionet.nsc.ru
The closing date for receiving extended abstracts of presentations and for registration is April 15, 2000.
The decision on acceptance for presentation will be communicated by May 15, 2000.
Example
A COMPUTER TOOL FOR INVESTIGATING EXTENDED REGULATORY REGIONS OF GENOMIC DNA SEQUENCES
Babenko V.N.*, Kosarev P.S., Vishnevsky O.V., Levitsky V.G., Basin V.V. and Frolov A.S.
Laboratory of Theoretical Genetics,
Institute of Cytology and Genetics,
Lavrentyev Ave., 10
Novosibirsk, 630090,
Russia
Phone: 7-(3832) 33-31-19
Fax: 7-(3832) 33-12-78
E.mail: bob@bionet.nsc.ru
Key words: regulatory regions, computer tool, repeats, nucleotide profile, oligonucleotides
Motivation: Despite the growing volume of the data on primary nucleotide sequences, the regulatory regions remain the major puzzle in their function. Numerous recognising programs considering a diversity of properties of regulatory regions have been developed. The system proposed here allows the specific contextual, conformational, and physico-chemical properties to be revealed basing on analysis of extended DNA regions.
Results: The Internet-accessible computer system RegScan designed to analyse the extended regulatory regions of eukaryotic genes has been developed. The computer system comprises the following software:
-
programs for classification dividing a set of promoters into TATA-containing and TATA-less promoters and promotes with and without CpG islands;
-
programs for constructing (a) nucleotide frequency profiles, (b) sequence complexity profiles, and (c) profiles of conformational and physico-chemical properties;
-
the program for constructing the sets of degenerate oligonucleotide motifs of a specified length;
-
the program searching for and visualising repeats in nucleotide sequences.
The system has allowed us to demonstrate the following characteristic patterns of vertebrate promoter regions: the TATA box region is flanked by regions with an increased G+C content and increased bending stiffness, the TATA box content is asymmetric, and promoter regions are saturated with both direct and inverted repeats.
Availability: The computer system RegScan is available via Internet at http://wwwmgs.bionet.nsc.ru/mgs/systems/regscan/
Introduction
Eukaryotic promoters are DNA sequences providing gene expression regulation at the stage of transcription initiation….
Methods and algorithms
Construction of position-specific nucleotide frequency profiles over the sample.
The construction of nucleotide frequency profiles is performed as follows. A set of phased sequences with a fixed length represents a nucleotide matrix. This matrix is decomposed into a series of successive overlapping windows (columns) of a given size. The number of nucleotides of a definite type Nia (i, the number of a window and a, designation of nucleotides in IUPAC 15 single letter-based code according to Cornish-Bowden (1985) is calculated within each window. The relative nucleotide frequency Nia/N (N is the total number of nucleotides in a window (column) of the nucleotide matrix) is plotted at the position corresponding to the window’s centre.
Implementation and results
We will illustrate operation of the system RegScan by computer analysis of vertebrate promoter sequences (Tabl. 1, Fig. 1).
Table 1. The number and percentage of promoters in samples
|
|
CpG+ |
CpG- |
|
|
TATA+ |
70 (23%) |
124 (41%) |
194 (64%) |
|
TATA- |
74 (25%) |
33 (11%) |
107 (36%) |
|
|
144 (48%) |
157 (52%) |
|
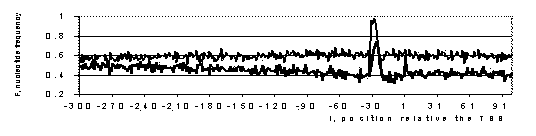
Figure 1. The frequency profiles of W(A+T) nucleotides for initial set of 301 promoters (bold line) and 301 false TATA-boxes (thin line). An increased concentration of these nucleotides is observed within the region with the center at –30 corresponding to TATA-box.
Discussion
We have developed a system for analysing extended regulatory regions of genomic DNA and applied it to study the vertebrate gene promoters transcribed by RNA polymerase II.
Acknowledgements
References
Barrick,D.,Villanueba,K., Childs,J., Kalil,R., Schneider,T.D., Lawrence,C.E., Gold,L. and Stormo,G.D. (1994) Quantitative analysis of ribosome binding sites in E.coli. Nucleic Acids Res., 22, 1287-1295.
Bendall,A.J., and Molloy,P.L., (1994) Base preferences for DNA binding by the bHLH-Zip protein USF: effects of MgCl2 on specificity and comparison with binding of Myc family members. Nucleic Acids Res., 22, 2801-2810.